
α-Ketoglutaric acid

| |
| Names | |
|---|---|
| Preferred IUPAC name
2-Oxopentanedioic acid | |
| Other names
2-Ketoglutaric acid
alpha-Ketoglutaric acid 2-Oxoglutaric acid Oxoglutaric acid | |
| Identifiers | |
3D model (JSmol)
|
|
| ChEBI | |
| ChemSpider | |
| DrugBank | |
| ECHA InfoCard | 100.005.756 |
| KEGG | |
| MeSH | alpha-ketoglutaric+acid |
PubChem CID
|
|
| UNII | |
CompTox Dashboard (EPA)
|
|
| |
| |
| Properties | |
| C5H6O5 | |
| Molar mass | 146.098 g·mol−1 |
| Melting point | 115 °C (239 °F; 388 K) |
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa).
| |
α-Ketoglutaric acid (also termed 2-oxoglutaric acid) is a dicarboxylic acid, i.e., a short-chain fatty acid containing two carboxyl groups (carboxy groups notated as CO2H) with C, O, and H standing for carbon, oxygen, and hydrogen, respectively (see adjacent figure). However, almost all animal tissues and extracellular fluids have a pH above 7. At these basic pH levels α-ketoglutaric acid exists almost exclusively as its conjugate base. That is, it has two negative electric charges due to its release of positively charged hydrogen (i.e., H+) from both of its now negatively charged carboxy groups, CO−2 (see Conjugate acid-base theory). This double negatively charged molecule is referred to as α-ketoglutarate or 2-oxoglutarate.[2]
β-Ketoglutaric acid (also termed 3-oxoglutaric acid and acetonedicarboxlic acid) and its conjugate base, β-Ketoglutarate, differ from α-ketoglutaric acid and α-ketoglutarate by the position of their ketone, i.e., carbon–oxygen double bond (C=O). β-Ketoglutaric acid's and β-ketoglutarate's C=O is on the second carbon from a CO2H whereas α-ketoglutaric acid's and α-ketoglutarate's C=O is on a carbon adjacent to a CO2H. "Ketoglutaric acid" and "ketoglutarate", when not qualified as α or β, almost always refers respectively to α-ketoglutaric acid or α-ketoglutarate.[2] β-Ketoglutarate does not have the biological actions that α-ketoglutarate has; it is even suggested to inhibit at least one action of α-ketoglutarate (see the following section titled, "β-Ketoglutaric acid and TET-2").[3] β-Ketoglutaric acid is used to synthesize other compounds (see applications of β-ketoglutaric acid) such as cyclohexenone which is itself widely used to synthesize other compounds.[4]
α-Ketoglutarate is an intermediate in the citric acid cycle; this cycle supplies the energy used by cells.[2] It is also an intermediate in or product of several other metabolic pathways.[2][5] These include its being a component of metabolic pathways that: make key amino acids and in the process regulate the cellular levels of carbon, nitrogen, and ammonia;[5] reduce the cellular levels of potentially toxic reactive oxygen species;[6][7] and synthesize the neurotransmitter gamma-aminobutyric acid.[8] It also acts as a direct stimulator of, or cofactor (i.e., required for but does not itself stimulate) for various cellular functions as defined in studies that are primarily preclinical (i.e., conducted in animal models of disease or on animal or human tissues). These studies have provided evidence that α-ketoglutarate contributes to regulating: kidney function;[9] the benefits that resistance exercise has in reducing obesity, strengthening muscles, and preventing muscle atrophy;[10] glucose tolerance as defined in glucose tolerance tests;[11] aging and the development of changes that are associated with aging including old age-related disorders and diseases;[12] the development and/or progression of certain types of cancer and inflammations;[13] and the differentiation of immature T cells into mature T cells.[14]
Functions
[edit]Metabolic interactions
[edit]Citric acid cycle
[edit]α-Ketoglutarate is a component of the citric acid cycle, a cyclical metabolic pathway located in the mitochondria. This cycle supplies the energy that cells need by sequentially metabolizing (indicated by →) citrate through seven intermediate metabolites and then converting the eighth intermediate metabolite, oxaloacetate, back to citrate:[2]
- citrate → cis-aconitate → isocitrate → α-ketoglutarate → succinyl-CoA → succinate → fumarate → malate → oxaloacetate → citrate
In this cycle, the enzyme isocitrate dehydrogenase 3 converts isocitrate (isocitrate has 4 isomers of which only the (−)-d-threo-isomer is the naturally occurring isomer in the citric acid cycle.[15]) to α-ketoglutarate which in the next step is converted to succinyl-CoA by the oxoglutarate dehydrogenase complex of enzymes. Outside of the citric acid cycle, α-ketoglutarate is made by a) the enzymes isocitrate dehydrogenase 1 or 2 which remove a carboxy group from isocitrate by oxidative decarboxylation to form α-ketoglutarate; b) glutaminolysis in which the enzyme glutaminase removes the amino group (i.e., −NH2) from glutamine to form glutamate which is converted to α-ketoglutarate by any one of three different enzymes, glutamate dehydrogenase, alanine transaminase, or aspartate transaminase (see The glutaminolytic pathways); and c) various pyridoxal phosphate-dependent transamination reactions mediated by, e.g., the alanine transaminase enzyme,[16] in which glutamate is converted to α-Ketoglutarate by "donating" its −NH2 to other compounds (see transamination).[5][17] Acting in these pathways, α-ketoglutarate contributes to the production of amino acids such as glutamine, proline, arginine, and lysine as well as the reduction of cellular carbon and nitrogen (i.e., N) levels; this prevents excessive levels of these two potentially toxic elements from accumulating in cells and tissues.[6][16][17] The neurotoxin, ammonia (i.e., NH3), is also prevented form accumulating in tissues. In this metabolic pathway the −NH2 group on an amino acid is transferred to α-ketoglutarate; this forms the α-keto acid of the original amino acid and the amine-containing product of α-ketoglutarate, glutamate. The celllular glutamate passes into the circulation and is taken up by the liver where it delivers its acquired −NH2 group to the urea cycle. In effect, the latter pathway removes excess ammonia from the body in the form of urinary urea.[6][7][18]
Reactive oxygen species
[edit]Many conditions can cause the excessive accumulation of reactive oxygen species such as the hydroxyl radical (i.e., •HO), hydrogen peroxide (i.e., H2O2), and superoxide anion (i.e., O2−). These tissue-injuring oxygen species may lead to excessive inflammation, atherosclerosis, cardiovascular diseases, neurological disorders, aging-associated diseases, and various cancers. Antioxidant enzymes (i.e., superoxide dismutase, catalase, and glutathione peroxidase) and non-enzymatic antioxidant agents (e.g., glutathione, vitamin C, and vitamin E) act to reduce the levels of these disease-causing agents. α-Ketoglutarate is one of the non-enzymatic antioxidant agents. It reacts with hydrogen peroxide (H2O2) to form succinate, carbon dioxide (i.e., CO2), and water (i.e., (H2O) thereby reducing the levels of H2O2. The protective action of α-ketoglutarate in reducing the toxic effects of H2O2 have been observed in Drosophila melanogaster (i.e., fruit flies), other animals, and humans. In addition, α-ketoglutarate increases the activity of superoxide dismutase which converts the highly toxic (O−
2) radical to molecular oxygen (i.e., O2) and H
2O
2.[6][7]
Formation of the neurotransmitter gamma-aminobutyric acid
[edit]A study conducted on the GABAergic neurons (i.e., nerve cells) in the neocortex of rat brains reported that the cytosolic form of the aspartate transaminase enzyme metabolizes α-ketoglutarate to glutamate which in turn is metabolized by glutamic acid decarboxylase to the inhibitory neurotransmitter gamma-aminobutyric acid. These metabolic reactions occur at the ends of the inhibitory axons of the GABAergic neurons and result in the release of gamma-aminobutyric acid which then inhibits the activation of nearby neurons.[8][19]
Bioactions of α-Ketoglutarate
[edit]OXGR1 receptor-dependent bioactions
[edit]OXGR1 (also known as GPR99) is a G protein-coupled receptor, i.e., a receptor located on the surface membrane of cells that binds certain ligands and is thereby stimulated to activate G proteins that elicit pre-programmed responses in their parent cells. OXRG1 was identified as a receptor for: a) α-ketoglutarate in 2004;[20][21] b) three leukotrienes viz., leukotrienes E4, C4, and D4 in 2013.[22][23] and c) itaconate in 2023.[20][21] These ligands have the following relative potencies in stimulating responses in OXGR1-bearing cells (Note that LTE4 can stimulate OXGR1 at concentrations far lower than those of the other four ligands):
- LTE4 >> LTC4 = LTD4 > α-ketoglutarate = itaconate.
It may be difficult to determine if an OXGR1-stimulating agent elicits a functional response by activating OXGR1 as opposed to some other mechanism. To make this distinction, studies have shown that the action of an OXGR1-activating agent on cultured cells, cultured tissues, or animals does not occur or is reduced when these cells, tissues, or animals have been altered so that they do not express or express greatly reduced levels of the OXGR1 protein,[20][21][22][24] or when their actions are inhibited by an OXGR1 receptor antagonists. OXGR1 is inhibited by Montelukast, a well-known inhibitor of the cysteinyl leukotriene receptor 1, i.e., the receptor for LTD4, LTC4, and LTE4. Montelukast also blocks the binding of these leukotrienes to, and thereby inhibits their activation of, OXGR1. One study presented evidence suggesting that α-ketoglutarate binds to OXGR1. It is assumed that Montelukast similarly blocks α-ketoglutarate's binding to, and thereby inhibits its activation of OXGR1.[22][24]
Kidney functions
[edit]The pendrin protein promotes the electroneutral exchange of tissue chloride (Cl−) for urinary bicarbonate (HCO3−) in the apical surfaces (i.e., surfaces facing the urine) of the kidney's renal β-intercalated cells (also termed type B intercalated cells) and non-α non-β intercalated cells (also termed non-A non-B intercalated cells) in the kidney's collecting duct system (i.e., CDS).[25] A study in mice found that OXGR1 colocalizes with pendrin in the β-intercalated cells and non-α non-β intercalated cells lining the tubules of their kidney's CDS. The intercalated cells in the CDS tubules isolated from mice used pendrin in cooperation with the electroneutral sodium bicarbonate exchanger 1 protein to mediate the Cl− for HCO3− exchange. α-Ketoglutarate stimulated the rate of this exchange in CDS tubules isolated from control mice (i.e., mice that had the Oxgr1 gene and protein) but not in CDS tubules isolated from Oxgr1 gene knockout mice (i.e., mice that lacked the Oxgr1 gene and protein). This study also showed that the α-ketoglutarate in the blood of mice filtered through their kidney's glomeruli into the proximal tubules and loops of Henle where it was reabsorbed. Mice drinking water with a basic pH (i.e., >7) due to the addition of sodium bicarbonate and mice lacking the Oxgr1 gene and protein who drink water without sodium bicarbonate had urines that were more basic (i.e., pH about 7.8) and contained higher levels of urinary α-ketoglutarate than control mice drinking water without this additive. Furthermore, Oxgr1 gene knockout mice drinking sodium bicarbonate-rich water developed metabolic alkalosis (body tissue pH levels higher than normal) that was associated with blood bicarbonate levels significantly higher and blood chloride levels significantly lower than those in control mice drinking the sodium bicarbonate-rich water.[9] Several other studies confirmed these findings and reported that cells in the proximal tubules of mice synthesize α-ketoglutarate and either broke it down thereby reducing its urine levels or secreted it into the tubules' lumens thereby increasing its urine levels.[26] Another study showed that a) In silico computer simulations strongly suggested that α-ketoglutarate bound to mouse OXGPR1; b) suspensions of canal duct cells isolated from the collecting ducts, loops of Henle, vasa recta, and interstitium of mouse kidneys raised their cytosolic ionic calcium, i.e., Ca2+ levels in response to α-ketoglutarate but this response (which is an indicator of cell activation) was blocked by pretreating the cells with Montelukast; and c) compared to mice not treated with streptozotocin, streptozotocin-induced diabetic mice (an animal disease model of diabetes) urinated only a small amount of the ionic sodium (Na+) that they drank or received by intravenous injections; Montelukast reversed this defect in the streptozotocin-pretreated mice.[24] These results indicate that in mice: a) α-ketoglutarate stimulates kidney OXGR1 to activate pendrin-mediated reabsorption of sodium and chloride by type B and non-A–non-B intercalated cells; b) high alkaline (i.e., sodium bicarbonate) intake produces significant increases in urine pH and α-ketoglutarate levels and impairs secretion of bicarbonate into the CDS tubules' lumens; c) the acid–base balance (i.e., levels of acids relative to their bases) in the face of high alkali intake depends on the activation of OXGR1 by α-ketoglutarate;[9][26] d) alkaline loading directly or indirectly stimulates α-ketoglutarate secretion into the kidney's proximal tubules where further down these tubules it activates OXGR1 and thereby the absorption and secretion of various agents that contribute to restoring a physiologically normal acid-base balance;[26] and e) α-ketoglutarate stimulates OXGR1-bearing CDS cells to raise their levels of cytosolic Ca2+) and in diabetic mice (and presumably other conditions involving high levels of blood and/or urine glucose) to increase these cells uptake of Na+.[9][24][25][26]
Resistance exercise, obesity, and muscle atrophy
[edit]Resistance exercise is exercising a muscle or muscle group against external resistance (see strength training). Studies have found that: a) mice feeding on a high fat or normal diet and given the resistance exercise of repeatedly climbing up a 1 meter ladder for 40 minutes had higher levels of α-ketoglutarate in their blood and 7 different muscles than non-exercising mice feeding respectively on the high fat or normal diet; b) mice conducting ladder climbing for several weeks and eating a high fat diet developed lower fat tissue masses and higher lean tissue masses than non-exercising mice on this diet; c) mice not in exercise training fed α-ketoglutarate likewise developed lower fat tissue and higher lean tissue masses than α-ketoglutarate-unfed, non-exercising mice; d) OXGR1 was strongly expressed in the mouse adrenal gland inner medullas and either resistance training or oral α-ketoglutarate increased this tissue's levels of the mRNA that is responsible for the synthesis of OXGR1; e) α-ketoglutarate stimulated chromaffin cells isolated from mouse adrenal glands to release epinephrine but reduction of these cells' OXGR1 levels by small interfering RNA reduced this response; f) α-ketoglutarate increased the blood serum levels of epinephrine in mice expressing OXGR1 but not in Oxgr1 gene knockout mice (i.e., mice lacking the OXGR1 gene and protein); g) mice on the high fat diet challenged with α-ketoglutarate increased their blood serum levels of epinephrine and developed lower fat tissue masses and higher lean tissue masses but neither OXGR1 gene knockout mice nor mice that had only their adrenal glands' OXGR1 gene knocked out showed these responses; and h) OXGR1 gene knockout mice fed the high fat diet developed muscle protein degradation, muscle atrophy (i.e., wasting), and falls in body weight whereas control mice did not show these fat diet-induced changes. These findings indicate that in mice resistance exercise increases muscle production as well as serum levels of α-ketoglutarate which in turn suppresses diet-induced obesity (i.e., low body fat and high lean body masses) at least in part by stimulating the OXGR1 on adrenal gland chromaffin cells to release epinephrine.[10][11][27] Another study reported that middle‐aged, i.e., 10‐month‐old, mice had lower serum levels of α-ketoglutarate than 2‐month‐old mice. Middle aged mice fed a high fat diet gained body weight and fat mass in the lower parts of their bodies and had impaired glucose tolerance as defined in glucose tolerance tests. Adding α-ketoglutarate to the drinking water of these mice inhibited the development of these changes. These results suggest that drinking the α-ketoglutarate-rich water replenished the otherwise diminished supplies of α-ketoglutarate in middle aged mice; the replenished supply of α-ketoglutarate thereby became available to suppress obesity and improve glucose tolerance.[28] Finally, a study in rats feed a low fat or high fat diet for 27 weeks and drinking α-ketoglutarate-rich water for the last 12 weeks of this 27 week period decreased their fat issue masses and increased their whole-body insulin sensitivity as defined in glucose tolerance tests. Rats fed either of these diets but not given α-ketoglutarate-rich water did not show these changes. This study indicates that α-ketoglutarate regulates body fat mass and insulin sensitivity in rats as well as mice.[29]
OXGR1 receptor-independent bioactions
[edit]The following actions of α-ketoglutarate have not been evaluated for their dependency on activating OXGR1 and are here assumed to be OXGR1-independent. Futures studies are needed to determine if OXGR1 contributes in whole or part to these actions of α-ketoglutarate.
Aging and diseases associated with aging
[edit]α-Ketoglutarate has been reported to increase the life span and/or delay the development of old age-related diseases in a species of roundworms and in mice. It nearly doubled the life span and delayed age-related deteriorations (e.g., decline in rapid, coordinated body movements) of Caenorhabditis elegans roundworms when added to their cell cultures.[5][30] Similarly, mice fed a diet high in calcium-bound α-ketoglutarate had a longer life span and shorter length of time in which they suffered old age-related morbidities (e.g., increased frailty, hair loss, and changes in body weight). Cell cultures of splenocytes (i.e., primarily T cells) from the α-ketoglutarate-fed mice produced higher levels of the anti-inflammatory cytokine, interleukin-10, than splenocytes from mice not fed α-ketoglutarate.[12][17] (Chronic low-grade inflammation which might be inhibited by interleukin-10, is associated with the development of old age-related disorders and diseases.[31])
A small and very preliminary study suggested that α-ketoglutarate may also promote longevity in humans. Fourteen females (age 64.09, range 43.49 to 72.46 years) and 28 males (age 62.78, range 41.31 to 79.57 years) volunteered to take Rejuvant® for an average period of 7 months. The Rejuvant® commercial preparations they used contained 1,000 mg of calcium α-ketoglutarate monohydrate plus either 900 mg of retinyl palmitate (a form of vitamin A containing 190 mg of calcium) for males (i.e., Rejuvant® for males) or 25 mg of vitamin D containing 190 mg of calcium for females (i.e., Rejuvant® for females).[32] As individuals age, their DNA develops additions of a methyl group (-CH3) to a cystine adjacent to a guanine (termed a CpG island) in an increasing number of CpG islands close to certain genes. These methylations often suppress the expression of the genes to which they are close. Assays (termed epigenetic clock tests) that determine the presence of methylations of cystines in CpG islands for key genes have been used to define an individual's biological age.[33][34][35] The Rejuvant® study reported that the median and range of the biological age of females before treatment was 62.15 (range, 46.4 to 73) years and fell to 55.55 (range 33.4 to 63.7) years after an average of 7 months treatment. These values for men were 61.85 (range 41.9 to 79.7) years before and 53.3 (33 to 74.9) years after treatment.[17][32] Overall, the combined group of males and females showed an average fall in biological age of 8 years compared to before treatment. The p-value for this difference was extraordinarily significant, i.e., 6.538x10-12, in showing that that this treatment decreased the participants' biological ages. However, the study did not: a) include a control group (i.e., concurrent study of individuals taking a placebo instead of Rejuvant®); b) determine if the retinyl palmitate, vitamin A, and/or calcium given with α-ketoglutarate contributed to the changes in biological ages; and c) disclose which genes were tracked for the methylation of their CpG island. The study recommended that studies need to include control groups taking a placebo or the appropriate dosages of retinyl palmitate, vitamin A, and calcium. Also, TruMe Labs, who were the maker and marketer of the biological age assay used in this study, sponsored part of the study and contributed three of its employees as authors to the study.[32]
Fe2+/α-ketoglutarate-dependent dioxygenase enzymes and TET enzymes
[edit]α-Ketoglutarate is a cofactor that is needed for certain enzymes in the histone-lysine demethylase protein superfamily to become activated. This superfamily consists of two groups, the FAD-dependent amine oxidases which do not require α-ketoglutarate for activation and the Fe2+/α-ketoglutarate-dependent dioxygenases (Fe2+ is the ferrous form of iron, i.e., Fe2+). The latter group of more than 30 enzymes is classified into 7 subfamilies termed histone lysine demethylases, i.e., HDM2 to HDM7, with each subfamily having multiple members. These HDMs are characterized by containing a Jumonji C (JmjC) protein domain. They function as dioxygenases or hydroxylases to remove methyl groups from the lysine residues on the histones enveloping DNA and thereby alter the expression of diverse genes.[36][37] These altered gene expressions lead to a wide range of changes in the functions of various cell types and thereby caused the development and/or progression of various cancers, pathological inflammations, and other disorders (see α-Ketoglutarate-dependent demethylase biological functions).[13][38] The TET enzymes (i.e., ten-eleven translocation (TET) methylcytosine dioxygenase family of enzymes) consists of three members, TET-1, TET-2, and TET-3. Like the Fe2+/α-ketoglutarate-dependent dioxygenases, all three TET enzymes require Fe2+ and α-ketoglutarate as cofactors to become activated. Unlike the dioxygenases, however, they remove methyl groups from the 5-methylcytosines of DNA sites that regulate the expression of nearby genes. These demethylations have a variety of effects including, similar to the Fe2+/α-ketoglutarate-dependent dioxygenases, alteration of the development and/or progression of various cancers, immune responses, and other disorders (see functions of TET enzymes).[39][40]
β-Ketoglutaric acid and TET-2
[edit]A recent study found that β-ketoglutaric acid was detected in the saliva of individuals chewing betel quid, a complex mixture derived from betel nuts mixed with various other materials. Chronic chewing betel quid is associated with the development of certain cancers, particularly those in the oral cavity. The study showed that β-ketoglutaric acid bound to the cancer-promoting protein TET-2 thereby inhibiting α-ketoglutarate's binding to this protein. Since α-ketoglutarate's binding of TET-2 is thought to be required for it to activate TET-2, the study suggested that β-ketoglutaric acid may not fulfill the requirements for TET-2 to be activatable and therefore may prove able to block α-ketoglutarate's cancer-promoting as well as inflammation-promoting and other actions that involve its activation of TET-2.[3]
Immune regulation
[edit]Under glutamine-deprived conditions, α-ketoglutarate promotes naïve CD4+ T cells differentiation into inflammation-promoting Th1 cells while inhibiting their differentiation into inflammation-inhibiting Treg cells thereby promoting certain inflammation responses.[14]
Interactive pathway map
[edit]Click on genes, proteins and metabolites below to link to respective articles. [§ 1]
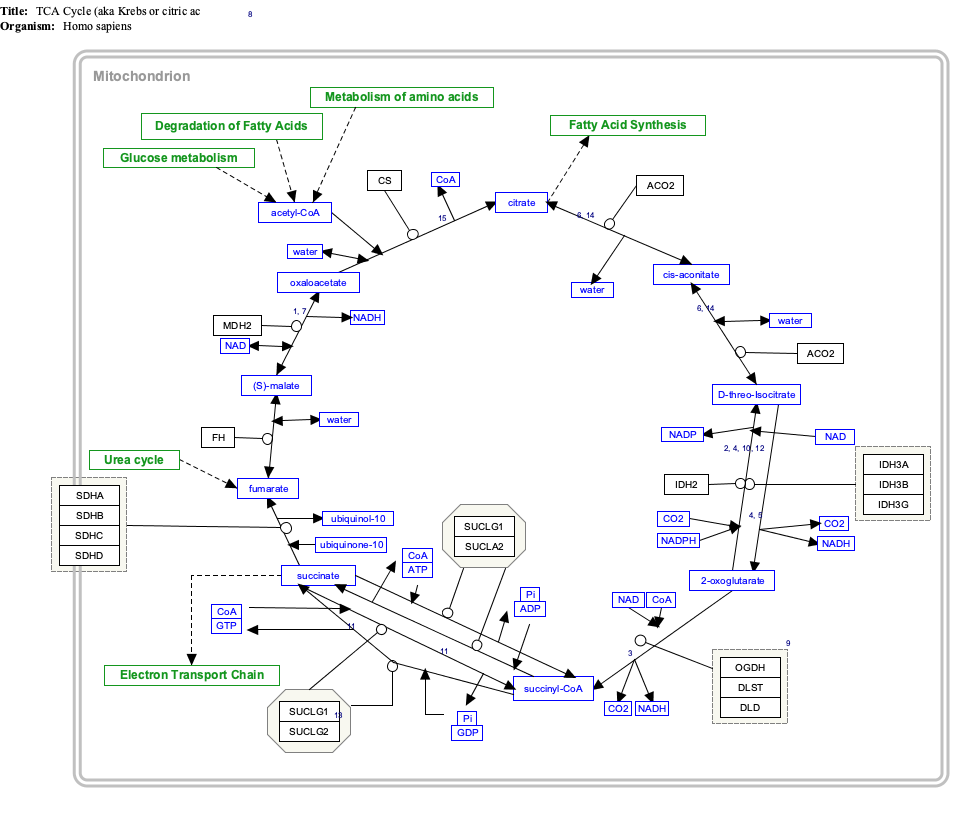
- ^ The interactive pathway map can be edited at WikiPathways: "TCACycle_WP78".
See also
[edit]References
[edit]- ^ Merck Index, 13th Edition, 5320.
- ^ a b c d e Chinopoulos C (August 2013). "Which way does the citric acid cycle turn during hypoxia? The critical role of α-ketoglutarate dehydrogenase complex". Journal of Neuroscience Research. 91 (8): 1030–43. doi:10.1002/jnr.23196. PMID 23378250.
- ^ a b Bhatkar D, Ananda N, Lokhande KB, Khunteta K, Jain P, Hebale A, Sarode SC, Sharma NK (September 2023). "Organic Acids Derived from Saliva-amalgamated Betel Quid Filtrate Are Predicted as a Ten-eleven Translocation-2 Inhibitor". Journal of Cancer Prevention. 28 (3): 115–130. doi:10.15430/JCP.2023.28.3.115. PMC 10564634. PMID 37830116.
- ^ Quintard A, Rodriguez J (June 2015). "Synergistic Cu-amine catalysis for the enantioselective synthesis of chiral cyclohexenones". Chemical Communications. 51 (46): 9523–6. doi:10.1039/c5cc02987b. PMID 25968341.
- ^ a b c d Wu N, Yang M, Gaur U, Xu H, Yao Y, Li D (January 2016). "Alpha-Ketoglutarate: Physiological Functions and Applications". Biomolecules & Therapeutics. 24 (1): 1–8. doi:10.4062/biomolther.2015.078. PMC 4703346. PMID 26759695.
- ^ a b c d Liu S, He L, Yao K (2018). "The Antioxidative Function of Alpha-Ketoglutarate and Its Applications". BioMed Research International. 2018: 3408467. doi:10.1155/2018/3408467. PMC 5884300. PMID 29750149.
- ^ a b c Kroupina K, Bémeur C, Rose CF (July 2022). "Amino acids, ammonia, and hepatic encephalopathy". Analytical Biochemistry. 649: 114696. doi:10.1016/j.ab.2022.114696. hdl:1866/26644. PMID 35500655.
- ^ a b Kaneko T, Mizuno N (August 1994). "Glutamate-synthesizing enzymes in GABAergic neurons of the neocortex: a double immunofluorescence study in the rat". Neuroscience. 61 (4): 839–49. doi:10.1016/0306-4522(94)90407-3. PMID 7838383.
- ^ a b c d Tokonami N, Morla L, Centeno G, Mordasini D, Ramakrishnan SK, Nikolaeva S, Wagner CA, Bonny O, Houillier P, Doucet A, Firsov D (July 2013). "α-Ketoglutarate regulates acid-base balance through an intrarenal paracrine mechanism". The Journal of Clinical Investigation. 123 (7): 3166–71. doi:10.1172/JCI67562. PMC 3696567. PMID 23934124.
- ^ a b Yuan Y, Xu P, Jiang Q, Cai X, Wang T, Peng W, Sun J, Zhu C, Zhang C, Yue D, He Z, Yang J, Zeng Y, Du M, Zhang F, Ibrahimi L, Schaul S, Jiang Y, Wang J, Sun J, Wang Q, Liu L, Wang S, Wang L, Zhu X, Gao P, Xi Q, Yin C, Li F, Xu G, Zhang Y, Shu G (April 2020). "Exercise-induced α-ketoglutaric acid stimulates muscle hypertrophy and fat loss through OXGR1-dependent adrenal activation". The EMBO Journal. 39 (7): e103304. doi:10.15252/embj.2019103304. PMC 7110140. PMID 32104923.
- ^ a b Yuan Y, Zhu C, Wang Y, Sun J, Feng J, Ma Z, Li P, Peng W, Yin C, Xu G, Xu P, Jiang Y, Jiang Q, Shu G (May 2022). "α-Ketoglutaric acid ameliorates hyperglycemia in diabetes by inhibiting hepatic gluconeogenesis via serpina1e signaling". Science Advances. 8 (18): eabn2879. Bibcode:2022SciA....8N2879Y. doi:10.1126/sciadv.abn2879. PMC 9067931. PMID 35507647.
- ^ a b Asadi Shahmirzadi A, Edgar D, Liao CY, Hsu YM, Lucanic M, Asadi Shahmirzadi A, Wiley CD, Gan G, Kim DE, Kasler HG, Kuehnemann C, Kaplowitz B, Bhaumik D, Riley RR, Kennedy BK, Lithgow GJ (September 2020). "Alpha-Ketoglutarate, an Endogenous Metabolite, Extends Lifespan and Compresses Morbidity in Aging Mice". Cell Metabolism. 32 (3): 447–456.e6. doi:10.1016/j.cmet.2020.08.004. PMC 8508957. PMID 32877690.
- ^ a b Manni W, Jianxin X, Weiqi H, Siyuan C, Huashan S (September 2022). "JMJD family proteins in cancer and inflammation". Signal Transduction and Targeted Therapy. 7 (1): 304. doi:10.1038/s41392-022-01145-1. PMC 9434538. PMID 36050314.
- ^ a b Klysz D, Tai X, Robert PA, Craveiro M, Cretenet G, Oburoglu L, Mongellaz C, Floess S, Fritz V, Matias MI, Yong C, Surh N, Marie JC, Huehn J, Zimmermann V, Kinet S, Dardalhon V, Taylor N (September 2015). "Glutamine-dependent α-ketoglutarate production regulates the balance between T helper 1 cell and regulatory T cell generation". Science Signaling. 8 (396): ra97. doi:10.1126/scisignal.aab2610. PMID 26420908.
- ^ Kamzolova SV, Shamin RV, Stepanova NN, Morgunov GI, Lunina JN, Allayarov RK, Samoilenko VA, Morgunov IG (2018). "Fermentation Conditions and Media Optimization for Isocitric Acid Production from Ethanol by Yarrowia lipolytica". BioMed Research International. 2018: 2543210. doi:10.1155/2018/2543210. PMC 5820659. PMID 29568744.
- ^ a b Yang RZ, Park S, Reagan WJ, Goldstein R, Zhong S, Lawton M, Rajamohan F, Qian K, Liu L, Gong DW (February 2009). "Alanine aminotransferase isoenzymes: molecular cloning and quantitative analysis of tissue expression in rats and serum elevation in liver toxicity". Hepatology. 49 (2): 598–607. doi:10.1002/hep.22657. PMC 2917112. PMID 19085960.
- ^ a b c d Gyanwali B, Lim ZX, Soh J, Lim C, Guan SP, Goh J, Maier AB, Kennedy BK (February 2022). "Alpha-Ketoglutarate dietary supplementation to improve health in humans". Trends in Endocrinology and Metabolism. 33 (2): 136–146. doi:10.1016/j.tem.2021.11.003. hdl:1871.1/4ada9cac-6437-44d5-ad2b-c0ee6431df3b. PMID 34952764.
- ^ Katayama, Kazuhiro (2004-12-01). "Ammonia metabolism and hepatic encephalopathy". Hepatology Research. 30: 73–80. doi:10.1016/j.hepres.2004.08.013. ISSN 1386-6346. PMID 15607143.
- ^ Robinson MB, Lee ML, DaSilva S (March 2020). "Glutamate Transporters and Mitochondria: Signaling, Co-compartmentalization, Functional Coupling, and Future Directions". Neurochemical Research. 45 (3): 526–540. doi:10.1007/s11064-020-02974-8. PMC 7060825. PMID 32002773.
- ^ a b c Zeng YR, Song JB, Wang D, Huang ZX, Zhang C, Sun YP, Shu G, Xiong Y, Guan KL, Ye D, Wang P (March 2023). "The immunometabolite itaconate stimulates OXGR1 to promote mucociliary clearance during the pulmonary innate immune response". The Journal of Clinical Investigation. 133 (6). doi:10.1172/JCI160463. PMC 10014103. PMID 36919698.
- ^ a b c Ye D, Wang P, Chen LL, Guan KL, Xiong Y (March 2024). "Itaconate in host inflammation and defense". Trends in Endocrinology and Metabolism. doi:10.1016/j.tem.2024.02.004. PMID 38448252.
- ^ a b c Kanaoka Y, Maekawa A, Austen KF (April 2013). "Identification of GPR99 protein as a potential third cysteinyl leukotriene receptor with a preference for leukotriene E4 ligand". The Journal of Biological Chemistry. 288 (16): 10967–72. doi:10.1074/jbc.C113.453704. PMC 3630866. PMID 23504326.
- ^ Sasaki F, Yokomizo T (August 2019). "The leukotriene receptors as therapeutic targets of inflammatory diseases". International Immunology. 31 (9): 607–615. doi:10.1093/intimm/dxz044. PMID 31135881.
- ^ a b c d Guerrero A, Visniauskas B, Cárdenas P, Figueroa SM, Vivanco J, Salinas-Parra N, Araos P, Nguyen QM, Kassan M, Amador CA, Prieto MC, Gonzalez AA (2021). "α-Ketoglutarate Upregulates Collecting Duct (Pro)renin Receptor Expression, Tubular Angiotensin II Formation, and Na+ Reabsorption During High Glucose Conditions". Frontiers in Cardiovascular Medicine. 8: 644797. doi:10.3389/fcvm.2021.644797. PMC 8220822. PMID 34179130.
- ^ a b Brazier F, Cornière N, Picard N, Chambrey R, Eladari D (April 2024). "Pendrin: linking acid base to blood pressure". Pflügers Archiv. 476 (4): 533–543. doi:10.1007/s00424-023-02897-7. PMID 38110744.
- ^ a b c d Grimm PR, Welling PA (September 2017). "α-Ketoglutarate drives electroneutral NaCl reabsorption in intercalated cells by activating a G-protein coupled receptor, Oxgr1". Current Opinion in Nephrology and Hypertension. 26 (5): 426–433. doi:10.1097/MNH.0000000000000353. PMID 28771454.
- ^ Cai X, Yuan Y, Liao Z, Xing K, Zhu C, Xu Y, Yu L, Wang L, Wang S, Zhu X, Gao P, Zhang Y, Jiang Q, Xu P, Shu G (January 2018). "α-Ketoglutarate prevents skeletal muscle protein degradation and muscle atrophy through PHD3/ADRB2 pathway". FASEB Journal. 32 (1): 488–499. doi:10.1096/fj.201700670R. PMC 6266637. PMID 28939592.
- ^ Tian Q, Zhao J, Yang Q, Wang B, Deavila JM, Zhu MJ, Du M (January 2020). "Dietary alpha-ketoglutarate promotes beige adipogenesis and prevents obesity in middle-aged mice". Aging Cell. 19 (1): e13059. doi:10.1111/acel.13059. PMC 6974731. PMID 31691468.
- ^ Tekwe CD, Yao K, Lei J, Li X, Gupta A, Luan Y, Meininger CJ, Bazer FW, Wu G (October 2019). "Oral administration of α-ketoglutarate enhances nitric oxide synthesis by endothelial cells and whole-body insulin sensitivity in diet-induced obese rats". Experimental Biology and Medicine. 244 (13): 1081–1088. doi:10.1177/1535370219865229. PMC 6775570. PMID 31357871.
- ^ Chin RM, Fu X, Pai MY, Vergnes L, Hwang H, Deng G, Diep S, Lomenick B, Meli VS, Monsalve GC, Hu E, Whelan SA, Wang JX, Jung G, Solis GM, Fazlollahi F, Kaweeteerawat C, Quach A, Nili M, Krall AS, Godwin HA, Chang HR, Faull KF, Guo F, Jiang M, Trauger SA, Saghatelian A, Braas D, Christofk HR, Clarke CF, Teitell MA, Petrascheck M, Reue K, Jung ME, Frand AR, Huang J (June 2014). "The metabolite α-ketoglutarate extends lifespan by inhibiting ATP synthase and TOR". Nature. 510 (7505): 397–401. doi:10.1038/nature13264. PMC 4263271. PMID 24828042.
- ^ Islam MT, Tuday E, Allen S, Kim J, Trott DW, Holland WL, Donato AJ, Lesniewski LA (February 2023). "Senolytic drugs, dasatinib and quercetin, attenuate adipose tissue inflammation, and ameliorate metabolic function in old age". Aging Cell. 22 (2): e13767. doi:10.1111/acel.13767. PMC 9924942. PMID 36637079.
- ^ a b c Demidenko O, Barardo D, Budovskii V, Finnemore R, Palmer FR, Kennedy BK, Budovskaya YV (November 2021). "Rejuvant®, a potential life-extending compound formulation with alpha-ketoglutarate and vitamins, conferred an average 8 year reduction in biological aging, after an average of 7 months of use, in the TruAge DNA methylation test". Aging. 13 (22): 24485–24499. doi:10.18632/aging.203736. PMC 8660611. PMID 34847066.
- ^ Soto-Palma C, Niedernhofer LJ, Faulk CD, Dong X (August 2022). "Epigenetics, DNA damage, and aging". The Journal of Clinical Investigation. 132 (16). doi:10.1172/JCI158446. PMC 9374376. PMID 35968782.
- ^ Chen L, Ganz PA, Sehl ME (2022). "DNA Methylation, Aging, and Cancer Risk: A Mini-Review". Frontiers in Bioinformatics. 2: 847629. doi:10.3389/fbinf.2022.847629. PMC 9580889. PMID 36304336.
- ^ Moqri M, Herzog C, Poganik JR, Justice J, Belsky DW, Higgins-Chen A, Moskalev A, Fuellen G, Cohen AA, Bautmans I, Widschwendter M, Ding J, Fleming A, Mannick J, Han JJ, Zhavoronkov A, Barzilai N, Kaeberlein M, Cummings S, Kennedy BK, Ferrucci L, Horvath S, Verdin E, Maier AB, Snyder MP, Sebastiano V, Gladyshev VN (August 2023). "Biomarkers of aging for the identification and evaluation of longevity interventions". Cell. 186 (18): 3758–3775. doi:10.1016/j.cell.2023.08.003. PMC 11088934. PMID 37657418.
- ^ Park SY, Park JW, Chun YS (March 2016). "Jumonji histone demethylases as emerging therapeutic targets". Pharmacological Research. 105: 146–51. doi:10.1016/j.phrs.2016.01.026. PMID 26816087.
- ^ Staehle HF, Pahl HL, Jutzi JS (December 2021). "The Cross Marks the Spot: The Emerging Role of JmjC Domain-Containing Proteins in Myeloid Malignancies". Biomolecules. 11 (12): 1911. doi:10.3390/biom11121911. PMC 8699298. PMID 34944554.
- ^ Maity J, Majumder S, Pal R, Saha B, Mukhopadhyay PK (November 2023). "Ascorbic acid modulates immune responses through Jumonji-C domain containing histone demethylases and Ten eleven translocation methylcytosine dioxygenase". BioEssays. 45 (11): e2300035. doi:10.1002/bies.202300035. PMID 37694689.
- ^ Joshi K, Liu S, Breslin SJ, Zhang J (June 2022). "Mechanisms that regulate the activities of TET proteins". Cellular and Molecular Life Sciences. 79 (7): 363. doi:10.1007/s00018-022-04396-x. PMC 9756640. PMID 35705880.
- ^ López-Moyado IF, Ko M, Hogan PG, Rao A (February 2024). "TET Enzymes in the Immune System: From DNA Demethylation to Immunotherapy, Inflammation, and Cancer". Annual Review of Immunology. 42. doi:10.1146/annurev-immunol-080223-044610. PMID 38360546.
| ||||||||||||










{kind=link}