
Electrophilic aromatic directing groups
In electrophilic aromatic substitution reactions, existing substituent groups on the aromatic ring influence the overall reaction rate or have a directing effect on positional isomer of the products that are formed.
An electron donating group (EDG) or electron releasing group (ERG, Z in structural formulas) is an atom or functional group that donates some of its electron density into a conjugated π system via resonance (mesomerism) or inductive effects (or induction)—called +M or +I effects, respectively—thus making the π system more nucleophilic.[1][2] As a result of these electronic effects, an aromatic ring to which such a group is attached is more likely to participate in electrophilic substitution reaction. EDGs are therefore often known as activating groups, though steric effects can interfere with the reaction.
An electron withdrawing group (EWG) will have the opposite effect on the nucleophilicity of the ring. The EWG removes electron density from a π system, making it less reactive in this type of reaction,[2][3] and therefore called deactivating groups.
EDGs and EWGs also determine the positions (relative to themselves) on the aromatic ring where substitution reactions are most likely to take place. Electron donating groups are generally ortho/para directors for electrophilic aromatic substitutions, while electron withdrawing groups (except the halogens) are generally meta directors. The selectivities observed with EDGs and EWGs were first described in 1892 and have been known as the Crum Brown–Gibson rule.[4]
Categories[edit]

Electron donating groups are typically divided into three levels of activating ability (The "extreme" category can be seen as "strong".) Electron withdrawing groups are assigned to similar groupings. Activating substituents favour electrophilic substitution about the ortho and para positions. Weakly deactivating groups direct electrophiles to attack the benzene molecule at the ortho- and para- positions, while strongly and moderately deactivating groups direct attacks to the meta- position.[5] This is not a case of favoring the meta- position like para- and ortho- directing functional groups, but rather disfavouring the ortho- and para-positions more than they disfavour the meta- position.
Activating groups[edit]
The activating groups are mostly resonance donors (+M). Although many of these groups are also inductively withdrawing (–I), which is a deactivating effect, the resonance (or mesomeric) effect is almost always stronger, with the exception of Cl, Br, and I.
| Magnitude of
activation |
Substituent Name (in approximate order
of activating strength) |
Structure | Type of electronic effect | Directing effect |
|---|---|---|---|---|
| Extreme | oxido group | -O− | +I, +M, metal-hydrogen exchange | ortho, para |
| Strong | (substituted) amino groups | -NH2, -NHR, -NR2 | –I, +M | |
| hydroxy and alkoxy groups | -OH,
-OR | |||
| Moderate | acylamido groups | -NHCOR | ||
| acyloxy groups | -OCOR | |||
| (di)alkylphosphino, alkylthio, and sulfhydryl groups[6] | -PR2,
-SR, -SH |
+M (weak) | ||
| Weak | phenyl (or aryl) group | -C6H5 | –I, +M;[7][8] though other interactions may be involved as well[9] | |
| vinyl group | -CH=CH2 | |||
| alkyl groups
(e.g. -CH3, -C2H5) |
-R | |||
| carboxylate group[10] | -CO2− | +I |
In general, the resonance effect of elements in the third period and beyond is relatively weak. This is mainly because of the relatively poor orbital overlap of the substituent's 3p (or higher) orbital with the 2p orbital of the carbon.
Due to a stronger resonance effect and inductive effect than the heavier halogens, fluorine is anomalous. The partial rate factor of electrophilic aromatic substitution on fluorobenzene is often larger than one at the para position, making it an activating group.[11] Conversely, it is moderately deactivated at the ortho and meta positions, due to the proximity of these positions to the electronegative fluoro substituent.
Deactivating groups[edit]
While all deactivating groups are inductively withdrawing (–I), most of them are also withdrawing through resonance (–M) as well. Halogen substituents are an exception: they are resonance donors (+M). With the exception of the halides, they are meta directing groups.
Halides are ortho, para directing groups but unlike most ortho, para directors, halides mildly deactivate the arene. This unusual behavior can be explained by two properties:
- Since the halogens are very electronegative they cause inductive withdrawal (withdrawal of electrons from the carbon atom of benzene).
- Since the halogens have non-bonding electrons they can donate electron density through pi bonding (resonance donation).
The inductive and resonance properties compete with each other but the resonance effect dominates for purposes of directing the sites of reactivity. For nitration, for example, fluorine directs strongly to the para position because the ortho position is inductively deactivated (86% para, 13% ortho, 0.6% meta). On the other hand, iodine directs to ortho and para positions comparably (54% para and 45% ortho, 1.3% meta).[12]
| Magnitude of
deactivation |
Substituent Name (in approximate order
of deactivating strength) |
Structure | Type of electronic effect | Directing effect |
|---|---|---|---|---|
| Strong | trifluoromethylsulfonyl group[13] | -SO2CF3 | –I, –M | meta |
| (substituted) ammonium groups[14] | -NR3+ (R = alkyl or H) | –I | ||
| nitro group | -NO2 | –I, –M | ||
| sulfonic acids and sulfonyl groups | -SO3H,
-SO2R | |||
| cyano group | -C≡N | |||
| trihalomethyl groups (strongest for -CF3 group) | -CX3 (X = F, Cl, Br, I) | –I | ||
| Moderate | haloformyl groups | -COX
(X = Cl, Br, I) |
–I, –M | |
| formyl and acyl groups | -CHO,
-COR | |||
| carboxyl and alkoxycarbonyl groups | -CO2H,
-CO2R | |||
| (substituted) aminocarbonyl groups | -CONH2,
-CONHR, -CONR2 | |||
| Weak | fluoro group (ortho, meta positions) | -F | –I, +M (ortho) | ortho, para |
| nitroso group | -N=O | –I, +M (dimer) or
–M (monomer) | ||
| halo groups | -F(para), -Cl, -Br, -I[15] | –I, +M (weak) |
Traditional rationalizations[edit]
Although the full electronic structure of an arene can only be computed using quantum mechanics, the directing effects of different substituents can often be guessed through analysis of resonance diagrams.

Specifically, any formal negative or positive charges in minor resonance contributors (ones in accord with the natural polarization but not necessarily obeying the octet rule) reflect locations having a larger or smaller density of charge in the molecular orbital for a bond most likely to break. A carbon atom with a larger coefficient will be preferentially attacked, due to more favorable orbital overlap with the electrophile.[16]
The perturbation of a conjugating electron-withdrawing or electron-donating group causes the π electron distribution on a benzene ring to resemble (very slightly!) an electron-deficient benzyl cation or electron-excessive benzyl anion, respectively. The latter species admit tractable quantum calculation using Hückel theory: the cation withdraws electron density at the ortho and para positions, favoring meta attack, whereas the anion releases electron density into the same positions, activating them for attack.[17] This is precisely the result that the drawing of resonance structures would predict.
For example, aniline has resonance structures with negative charges around the ring system:

Attack occurs at ortho and para positions, because the (partial) formal negative charges at these positions indicate a local electron excess. On the other hand, the nitrobenzene resonance structures have positive charges around the ring system:

Attack occurs at the meta position, since the (partial) formal positive charges at the ortho and para positions indicate electron deficiency at these positions.
Another common argument, which makes identical predictions, considers the stabilization or destabilization by substituents of the Wheland intermediates resulting from electrophilic attack at the ortho/para or meta positions. The Hammond postulate then dictates that the relative transition state energies will reflect the differences in the ground state energies of the Wheland intermediates.[14][18]
Carbonyls, sulfonic acids and nitro[edit]
Because of the full or partial positive charge on the element directly attached to the ring for each of these groups, they all have a moderate to strong electron-withdrawing inductive effect (known as the -I effect). They also exhibit electron-withdrawing resonance effects, (known as the -M effect):
Thus, these groups make the aromatic ring very electron-poor (δ+) relative to benzene and, therefore, they strongly deactivate the ring (i.e. reactions proceed much slower in rings bearing these groups compared to those reactions in benzene.)
Anilines, phenols, and ethers (such as anisole)[edit]
Due to the electronegativity difference between carbon and oxygen / nitrogen, there will be a slight electron withdrawing effect through inductive effect (known as the –I effect). However, the other effect called resonance add electron density back to the ring (known as the +M effect) and dominate over that of inductive effect. Hence the result is that they are EDGs and ortho/para directors.
Phenol is an ortho/para director, but in a presence of base, the reaction is more rapid. It is due to the higher reactivity of phenolate anion. The negative oxygen was 'forced' to give electron density to the carbons (because it has a negative charge, it has an extra +I effect). Even when cold and with neutral (and relatively weak) electrophiles, the reaction still occurs rapidly.

Alkyl groups[edit]
Alkyl groups are electron donating groups. The carbon on that is sp3 hybridized and less electronegative than those that are sp2 hybridized. They have overlap on the carbon–hydrogen bonds (or carbon–carbon bonds in compounds like tert-butylbenzene) with the ring p orbital. Hence they are more reactive than benzene and are ortho/para directors.
Carboxylate[edit]
Inductively, the negatively charged carboxylate ion moderately repels the electrons in the bond attaching it to the ring. Thus, there is a weak electron-donating +I effect. There is an almost zero -M effect since the electron-withdrawing resonance capacity of the carbonyl group is effectively removed by the delocalisation of the negative charge of the anion on the oxygen. Thus overall the carboxylate group (unlike the carboxyl group) has an activating influence.[10]

Alkylammonium and trifluoromethyl group[edit]
These groups have a strong electron-withdrawing inductive effect (-I) either by virtue of their positive charge or because of the powerfully electronegativity of the halogens. There is no resonance effect because there are no orbitals or electron pairs which can overlap with those of the ring. The inductive effect acts like that for the carboxylate anion but in the opposite direction (i.e. it produces small positive charges on the ortho and para positions but not on the meta position and it destabilises the Wheland intermediate.) Hence these groups are deactivating and meta directing:

Halides' competing effects[edit]
Induction versus resonance[edit]
Fluorine is something of an anomaly in this circumstance. Above, it is described as a weak electron withdrawing group but this is only partly true. It is correct that fluorine has a -I effect, which results in electrons being withdrawn inductively. However, another effect that plays a role is the +M effect which adds electron density back into the benzene ring (thus having the opposite effect of the -I effect but by a different mechanism). This is called the mesomeric effect (hence +M) and the result for fluorine is that the +M effect approximately cancels out the -I effect. The effect of this for fluorobenzene at the para position is reactivity that is comparable to (or even higher than) that of benzene. Because inductive effects depends strongly on proximity, the meta and ortho positions of fluorobenzene are considerably less reactive than benzene. Thus, electrophilic aromatic substitution on fluorobenzene is strongly para selective.
This -I and +M effect is true for all halides - there is some electron withdrawing and donating character of each. To understand why the reactivity changes occur, we need to consider the orbital overlaps occurring in each. The valence orbitals of fluorine are the 2p orbitals which is the same for carbon - hence they will be very close in energy and orbital overlap will be favourable. Chlorine has 3p valence orbitals, hence the orbital energies will be further apart and the geometry less favourable, leading to less donation the stabilize the carbocationic intermediate, hence chlorobenzene is less reactive than fluorobenzene. However, bromobenzene and iodobenzene are about the same or a little more reactive than chlorobenzene, because although the resonance donation is even worse, the inductive effect is also weakened due to their lower electronegativities. Thus the overall order of reactivity is U-shaped, with a minimum at chlorobenzene/bromobenzene (relative nitration rates compared to benzene = 1 in parentheses): PhF (0.18) > PhCl (0.064) ~ PhBr (0.060) < PhI (0.12).[12] But still, all halobenzenes reacts slower than benzene itself.
Notice that iodobenzene is still less reactive than fluorobenzene because polarizability plays a role as well. This can also explain why phosphorus in phosphanes can't donate electron density to carbon through induction (i.e. +I effect) although it is less electronegative than carbon (2.19 vs 2.55, see electronegativity list) and why hydroiodic acid (pKa = -10) being much more acidic than hydrofluoric acid (pKa = 3). (That's 1013 times more acidic than hydrofluoric acid)
Directing effect[edit]
Due to the lone pair of electrons, halogen groups are available for donating electrons. Hence they are therefore ortho / para directors.
Nitroso group[edit]
Induction[edit]
Due to the electronegativity difference between carbon and nitrogen, the nitroso group has a relatively strong -I effect, but not as strong as the nitro group. (Positively charged nitrogen atoms on alkylammonium cations and on nitro groups have a much stronger -I effect)
Resonance[edit]
The nitroso group has both a +M and -M effect, but the -M effect is more favorable.
Nitrogen has a lone pair of electrons. However, the lone pair of its monomer form is unfavourable to donate through resonance. Only the dimer form is available for +M effect. However, the dimer form is less stable in a solution. Therefore, the nitroso group is less available to donate electrons.
Oppositely, withdrawing electron density is more favourable: (see the picture on the right).
.jpg)
As a result, the nitroso group is a deactivator. However, it has available to donate electron density to the benzene ring during the Wheland intermediate, making it still being an ortho / para director.
Steric effects[edit]
There are 2 ortho positions, 2 meta positions and 1 para position on benzene when a group is attached to it. When a group is an ortho / para director with ortho and para positions reacting with the same partial rate factor, we would expect twice as much ortho product as para product due to this statistical effect. However, the partial rate factors at the ortho and para positions are not generally equal. In the case of a fluorine substituent, for instance, the ortho partial rate factor is much smaller than the para, due to a stronger inductive withdrawal effect at the ortho position. Aside from these effects, there is often also a steric effect, due to increased steric hindrance at the ortho position but not the para position, leading to a larger amount of the para product.
The effect is illustrated for electrophilic aromatic substitutions with alkyl substituents of differing steric demand for electrophilic aromatic nitration.[19]
| Substrate | toluene [-CH3] | ethylbenzene
[-CH2CH3] |
cumene
[-CH(CH3)2] |
tert-butylbenzene
[-C(CH3)3] |
|---|---|---|---|---|
| ortho product | 58 | 45 | 30 | 16 |
| meta product | 5 | 6 | 8 | 11 |
| para product | 37 | 59 | 62 | 73 |
| ortho/para ratio | 1.57 | 0.76 | 0.48 | 0.22 |
The methyl group in toluene is small and will lead the ortho product being the major product. On the other hand, the t-butyl group is very bulky (there are 3 methyl groups attached to a single carbon) and will lead the para product as the major one. Even with toluene, the product is not 2:1 but having a slightly less ortho product.
Directing effect on multiple substituents[edit]
When two substituents are already present on the ring, the third substituent's new location is relatively predictable. If the existing substituents reinforce or the molecule is highly symmetric, there may be no ambiguity. Otherwise:[20]
- The most-activating substituent usually controls over the less-activating one.

Substituents add ortho to the amine in diethyl-(para-methyl)aniline and ortho to the amide in para-cyanobenzamide - In particular, ortho/para directors control over meta ones.

Substituents add ortho to the amine in diethyl-(meta-trifluoromethyl)aniline and ortho to the fluoride in para-fluorobenzaldehyde - When multiple substituents are comparably activating, steric hindrance dominates regioselectivity.

Substituents add ortho to the methyl group in para-(tert-butyl)toluene - In particular, the position between two substituents, each meta to the other, reacts last.
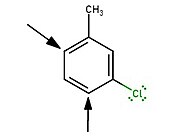
New substituents add para to either substituent in meta-chlorotoluene




See also[edit]
References[edit]
- ^ "Electron withdrawing group". Illustrated Glossary of Organic Chemistry. UCLA Department of Chemistry. Retrieved 16 November 2012.
- ^ a b Hunt, Ian. "Substituent Effects". University of Calgary Department of Chemistry. Retrieved 16 November 2012.
- ^ "Electron donating group". Illustrated Glossary of Organic Chemistry. UCLA Department of Chemistry. Retrieved 16 November 2012.
- ^ Brown, A. Crum; Gibson, John (1892). "XXX.—A rule for determining whether a given benzene mono-derivative shall give a meta-di-derivative or a mixture of ortho- and para-di-derivatives". J. Chem. Soc. 61: 367–369. doi:10.1039/ct8926100367.
- ^ "Substituent Effects". www.mhhe.com. Retrieved 2 April 2015.
- ^ James, Ashenhurst (29 Jan 2018). "Ortho-, Para- and Meta- Directors in Electrophilic Aromatic Substitution". Master Organic Chemistry.
- ^ Norman, Richard O. C.; Coxon, James M. (1993). Principles of Organic Synthesis (3rd ed.). CRC Press. pp. 353–354. ISBN 9780748761623.
- ^ Salvatella, Luis (October 2017). "The alkyl group is a –I + R substituent". Educación Química. 28 (4): 232–237. doi:10.1016/j.eq.2017.06.004. hdl:10261/184773. S2CID 46641895.
- ^ Hoggett, J. G.; Moodie, R. B.; Penton, J. R.; Schofield, K. (1971). Nitration and aromatic reactivity. London: Cambridge University Press. p. 200. ISBN 0521080290. OCLC 205846.
- ^ a b Smith, Ed (12 February 2018). "LECTURE 2" (PDF). Handouts for Organic Chemistry Lectures given at Imperial College London, Chemistry. p. 3.
- ^ Rosenthal, Joel; Schuster, David I. (2003-06-01). "The Anomalous Reactivity of Fluorobenzene in Electrophilic Aromatic Substitution and Related Phenomena". Journal of Chemical Education. 80 (6): 679–690. Bibcode:2003JChEd..80..679R. doi:10.1021/ed080p679. ISSN 0021-9584.
- ^ a b Jonathan., Clayden (2012). Organic chemistry. Greeves, Nick., Warren, Stuart G. (2nd ed.). Oxford: Oxford University Press. ISBN 9780199270293. OCLC 761379371.
- ^ Andrew, D. Abell; Brent, K. Nabbs; Alan, R. Battersby (12 February 1998). "Synthesis and Properties of Ring-Deactivated Deuterated (Hydroxymethyl)pyrroles". Journal of the American Chemical Society. 120 (8). doi:10.1021/ja973656+.
- ^ a b C., Vollhardt, K. Peter (2018-01-29). Organic chemistry : structure and function. Schore, Neil Eric, 1948- (8e ed.). New York. ISBN 9781319079451. OCLC 1007924903.
{{cite book}}: CS1 maint: location missing publisher (link) CS1 maint: multiple names: authors list (link) - ^ "Substitution Reactions of Benzene Derivatives". Chemistry LibreTexts. 2013-10-02. Retrieved 2021-09-18.
- ^ E., Lewis, David (2016). Advanced organic chemistry. New York. ISBN 9780199758975. OCLC 933277973.
{{cite book}}: CS1 maint: location missing publisher (link) CS1 maint: multiple names: authors list (link) - ^ Fleming, Ian (1976). Frontier orbitals and organic chemical reactions. London: Wiley. ISBN 0471018201. OCLC 2048204.
- ^ Carey, Francis A. (2013-01-07). Organic chemistry. Giuliano, Robert M., 1954- (Ninth ed.). New York, NY. ISBN 9780073402741. OCLC 822971422.
{{cite book}}: CS1 maint: location missing publisher (link) - ^ Peter, Sykes (1979). "2" (PDF). Some Organic Reaction Pathways. p. 32. ISBN 0851869998.
- ^ "12.15. Multiple Multiple Substituent Substituent Effects" (PDF). p. 7.