
α,β-Unsaturated carbonyl compound

R2 & R4 can also be single hydrogens.
α,β-Unsaturated carbonyl compounds are organic compounds with the general structure (O=CR)−Cα=Cβ-R.[1][2] Such compounds include enones and enals, but also carboxylic acids and the corresponding esters and amides. In these compounds, the carbonyl group is conjugated with an alkene (hence the adjective unsaturated). Unlike the case for carbonyls without a flanking alkene group, α,β-unsaturated carbonyl compounds are susceptible to attack by nucleophiles at the β-carbon. This pattern of reactivity is called vinylogous. Examples of unsaturated carbonyls are acrolein (propenal), mesityl oxide, acrylic acid, and maleic acid. Unsaturated carbonyls can be prepared in the laboratory in an aldol reaction and in the Perkin reaction.
Classifications[edit]
α,β-Unsaturated carbonyl compounds can be subclassified according to the nature of the carbonyl and alkene groups.
- Parent α,β-Unsaturated Carbonyls
-
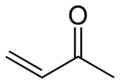 Methyl vinyl ketone, the simplest α,β-unsaturated ketone
Methyl vinyl ketone, the simplest α,β-unsaturated ketone -
 Acrolein, the simplest α,β-unsaturated aldehyde
Acrolein, the simplest α,β-unsaturated aldehyde -
 Methyl acrylate, an α,β-unsaturated ester
Methyl acrylate, an α,β-unsaturated ester -
 Acrylamide, precursor to polyacrylamide
Acrylamide, precursor to polyacrylamide -
 Maleic acid, an α,β-unsaturated dicarbonyl
Maleic acid, an α,β-unsaturated dicarbonyl -
 Fumaric acid, isomeric with maleic acid
Fumaric acid, isomeric with maleic acid -

-








Acryloyl group[edit]

α,β-Unsaturated carbonyl compounds featuring a carbonyl conjugated to an alkene that is terminal, or vinylic, contain the acryloyl group (H2C=CH−C(=O)−); it is the acyl group derived from acrylic acid. The preferred IUPAC name for the group is prop-2-enoyl, and it is also known as acrylyl or simply (and incorrectly) as acryl. Compounds containing an acryloyl group can be referred to as "acrylic compounds".
α,β-Unsaturated acids, esters, and amides[edit]
An α,β-unsaturated acid is a type of α,β-unsaturated carbonyl compound that consists of an alkene conjugated to a carboxylic acid.[3] The simplest example is acrylic acid (CH2=CHCO2H). These compounds are prone to polymerization, giving rise to the large area of polyacrylate plastics. Acrylate polymers are derived from but do not contain the acrylate group.[4] The carboxyl group of acrylic acid can react with ammonia to form acrylamide, or with an alcohol to form an acrylate ester. Acrylamide and methyl acrylate are commercially important examples of α,β-unsaturated amides and α,β-unsaturated esters, respectively. They also polymerize readily. Acrylic acid, its esters, and its amide derivatives feature the acryloyl group.
α,β-Unsaturated dicarbonyls are also common. The parent compounds are maleic acid and the isomeric fumaric acid. Maleic acid forms esters, an imide, and an anhydride, i.e. diethyl maleate, maleimide, and maleic anhydride. Fumaric acid, as fumarate, is an intermediate in the Krebs citric acid cycle, which is of great importance in bioenergy.
Enones[edit]
An enone (or alkenone) is an organic compound containing both alkene and ketone functional groups. In an α,β-unsaturated enone, the alkene is conjugated to the carbonyl group of the ketone.[3] The simplest enone is methyl vinyl ketone (butenone, CH2=CHCOCH3). Enones are typically produced using an aldol condensation or Knoevenagel condensation. Some commercially significant enones produced by condensations of acetone are mesityl oxide (dimer of acetone) and phorone and isophorone (trimers).[5] In the Meyer–Schuster rearrangement, the starting compound is a propargyl alcohol. Another method to access α,β-unsaturated carbonyls is via selenoxide elimination. Cyclic enones can be prepared via the Pauson–Khand reaction.

Cyclic enones[edit]
The cyclic enones include cyclopropenone, cyclobutenone,[6] cyclopentenone, cyclohexenone, and cycloheptenone.[7]
Enals[edit]
An enal (or alkenal) is an organic compound containing both alkene and aldehyde functional groups. In an α,β-unsaturated enal, the alkene is conjugated to the carbonyl group of the aldehyde (formyl group).[3] The simplest enal is acrolein (CH2=CHCHO). Other examples include cis-3-hexenal (essence of mowed lawns) and cinnamaldehyde (essence of cinnamon).
- Other α,β-Unsaturated Carbonyls
-
 E-Crotonaldehyde, an enal that exists as an isomer
E-Crotonaldehyde, an enal that exists as an isomer -
 Cyclohexenone, common cyclic enone
Cyclohexenone, common cyclic enone -
 testosterone, the male sex hormone
testosterone, the male sex hormone -
 Cinnamaldehyde, essence of cinnamon
Cinnamaldehyde, essence of cinnamon -
 Paraquinone, a particularly electrophilic α,β-unsaturated carbonyl
Paraquinone, a particularly electrophilic α,β-unsaturated carbonyl -
 Enone complex of iron tricarbonyl
Enone complex of iron tricarbonyl -




iron-tricarbonyl-2D-skeletal.png)

Reactions of α,β-unsaturated carbonyls[edit]
α,β-Unsaturated carbonyls are electrophilic at both the carbonyl carbon as well as the β-carbon. Depending on conditions, either site is attacked by nucleophiles. Additions to the alkene are called conjugate additions. One type of conjugate addition is the Michael addition, which is used commercially in the conversion of mesityl oxide into isophorone. Owing to their extended conjugation, α,β-unsaturated carbonyls are prone to polymerization. In terms of industrial scale, polymerization dominates the use of α,β-unsaturated carbonyls. Again because of their electrophilic character, the alkene portion of α,β-unsaturated carbonyls is good dienophiles in Diels–Alder reactions. They can be further activated by Lewis acids, which bind to the carbonyl oxygen. α,β-Unsaturated carbonyls are good ligands for low-valent metal complexes, examples being Fe(bda)(CO)3 and tris(dibenzylideneacetone)dipalladium(0).
α,β-Unsaturated carbonyls are readily hydrogenated. Hydrogenation can target the carbonyl or the alkene (conjugate reduction) selectively, or both functional groups.
Enones undergo the Nazarov cyclization reaction and in the Rauhut–Currier reaction (dimerization).
α,β-Unsaturated thioesters[edit]
α,β-Unsaturated thioesters are intermediates in several enzymatic processes. Two prominent examples are coumaroyl-coenzyme A and crotonyl-coenzyme A. They arise by the action of acyl-CoA dehydrogenases.[8] Flavin adenine dinucleotide (FAD) is a required co-factor.

Safety[edit]
Since α,β-unsaturated compounds are electrophiles and alkylating agents, many α,β-unsaturated carbonyl compounds are toxic. The endogenous scavenger compound glutathione naturally protects from toxic electrophiles in the body. Some drugs (amifostine, N-acetylcysteine) containing thiol groups may protect from such harmful alkylation.
See also[edit]
References[edit]
- ^ Patai, Saul; Rappoport, Zvi, eds. (1989). Enones: Vol. 1 (1989). Patai's Chemistry of Functional Groups. doi:10.1002/9780470772218. ISBN 9780470772218.
- ^ Patai, Saul; Rappoport, Zvi, eds. (1989). Enones: Vol. 2 (1989). Patai's Chemistry of Functional Groups. doi:10.1002/9780470772225. ISBN 9780470772225.
- ^ a b c Smith, Michael B.; March, Jerry (2007), Advanced Organic Chemistry: Reactions, Mechanisms, and Structure (6th ed.), New York: Wiley-Interscience, ISBN 978-0-471-72091-1
- ^ Ohara, Takashi; Sato, Takahisa; Shimizu, Noboru; Prescher, Günter; Schwind, Helmut; Weiberg, Otto; Marten, Klaus; Greim, Helmut (2003). "Acrylic Acid and Derivatives". Ullmann's Encyclopedia of Industrial Chemistry. Weinheim: Wiley-VCH. doi:10.1002/14356007.a01_161.pub2. ISBN 978-3527306732.
- ^ Siegel, Hardo; Eggersdorfer, Manfred (2000). "Ketones". Ullmann's Encyclopedia of Industrial Chemistry. Weinheim: Wiley-VCH. doi:10.1002/14356007.a15_077. ISBN 978-3527306732.
- ^ Ross, A. G.; Li, X.; Danishefsky, S. J. (2012). "Preparation of Cyclobutenone". Organic Syntheses. 89: 491. doi:10.15227/orgsyn.089.0491.
- ^ Ito, Y.; Fujii, S.; Nakatuska, M.; Kawamoto, F.; Saegusa, T. (1979). "One-Carbon Ring Expansion of Cycloalkanones to Conjugated Cycloalkenones: 2-Cyclohepten-1-One". Organic Syntheses. 59: 113. doi:10.15227/orgsyn.059.0113.
- ^ Thorpe, Colin; Kim, Jujng-Ja P. (1 June 1995). "Structure and mechanism of action of the Acyl-CoA dehydrogenases". The FASEB Journal. 9 (9): 718–725. doi:10.1096/fasebj.9.9.7601336. ISSN 0892-6638. PMID 7601336. S2CID 42549744.