
Carbometalation

A carbometallation is any reaction where a carbon-metal bond reacts with a carbon-carbon π-bond to produce a new carbon-carbon σ-bond and a carbon-metal σ-bond.[1] The resulting carbon-metal bond can undergo further carbometallation reactions (oligomerization or polymerization see Ziegler-Natta polymerization) or it can be reacted with a variety of electrophiles including halogenating reagents, carbonyls, oxygen, and inorganic salts to produce different organometallic reagents. Carbometallations can be performed on alkynes and alkenes to form products with high geometric purity or enantioselectivity, respectively. Some metals prefer to give the anti-addition product with high selectivity and some yield the syn-addition product. The outcome of syn and anti- addition products is determined by the mechanism of the carbometallation.
Carboboration
[edit]Carboboration is one of the most versatile carbometallation reactions. See Carboboration.
Carboalumination
[edit]The carboalumination reaction is most commonly catalyzed by zirconocene dichloride (or related catalyst). Some carboaluminations are performed with titanocene complexes.[1] This reaction is sometimes referred to as the Zr- catalyzed asymmetric carboalumination of alkenes (ZACA) or the Zr-catalyzed methylalumination of alkynes (ZMA).[2]
.png)
The most common trialkyl aluminium reagents for this transformation are trimethylaluminium, triethylaluminium, and sometimes triisobutylaluminium. When using trialkylaluminium reagents that have beta-hydrides, eliminations and hydroaluminium reactions become competing processes. The general mechanism of the ZMA reaction can be described as first the formation of the active catalytic species from the pre-catalyst zirconocene dichloride through its reaction with trimethyl aluminium. First transmetalation of a methyl from the aluminium to the zirconium occurs. Next, chloride abstraction by aluminium creates a cationic zirconium species that is closely associated with an anionic aluminium complex. This zirconium cation can coordinate an alkene or alkyne where migratory insertion of a methyl then takes place. The resultant vinyl or alkyl zirconium species can undergo a reversible, but stereoretentive transmetalation with an organoaluminium to provide the carboalumination product and regeneration of the zirconocene dichloride catalyst. This process generally provides the syn-addition product; however, conditions exist to provide the anti-addition product though a modified mechanism.
Trimethylsilyl (TMS) protected alkynes, trimethyl germanium alkynes, and terminal alkynes can produce anti-carboalumination products at room temperature or elevated temperatures if a coordinating group is nearby on the substrate.[3] In these reactions, first syn-carboalumination takes place under the previously outlined mechanism. Then, another equivalent of aluminium that is coordinated to the directing group can displace the vinyl aluminium, inverting the geometry at the carbon where displacement takes place.

This forms a thermodynamically favorable metallacycle to prevent subsequent inversions. Formally, this process provides anti-carboalumination products that can be quenched with electrophiles. A limitation of this methodology is that the directing group must be sufficiently close to the carbon-carbon π-bond to form a thermodynamically favorable ring or else mixtures of geometric isomers will form.

The carboalumination of alkenes to form substituted alkanes can be rendered enantioselective if prochiral alkenes are used. In these reactions, a chiral indenyl zirconium catalyst is used to induce enantioselectivity. In these reactions, high enantioselectivities were obtained for several trialkyl aluminium reagents, however, the yield decreases dramatically with each additional carbon of the alkyl chain on the trialkyl aluminium reagent.[2]
Carbolithiation
[edit]
Carbolithiation is the addition of an organolithium reagent across a carbon-carbon pi-bond. The organolithium reagents used in this transformation can be commercial (such as n-butyllithium) or can be generated through deprotonation or lithium-halogen exchange.[4][5] Both inter- and intramolecular examples of carbolithiation exist and can be used in synthesis to generate complexity. Organolithiums are highly reactive chemicals and often the resulting organolithium reagent generated from the carbolithiation can continue to react with electrophiles or remaining starting material (resulting in polymerization).[4] This reaction has been rendered enantioselective[6][7] through the use of sparteine, which can chelate the lithium ion and induce chirality.[4] Today, this is not a common strategy due to a shortage of natural sparteine. However, recent advances in the synthesis of sparteine surrogates and their effective application in carbolithiation have reactivated interest in this strategy.[8]
Another demonstration of this reaction type is an alternative route to tamoxifen starting from diphenylacetylene and ethyllithium:[9] The capturing electrophile here is triisopropyl borate forming the boronic acid R–B(OH)2. The second step completing tamoxifen is a Suzuki reaction.

Tamoxifen carbometalation

As a consequence of the high reactivity of organolithiums as strong bases and strong nucleophiles, the substrate scope of the carbolithiation is generally limited to chemicals that do not contain acidic or electrophilic functional groups.
Carbomagnesiation and carbozincation
[edit]Due to the decreased nucleophilicity of Grignard reagents (organomagnesium) and organozinc reagents, non-catalyzed carbomagnesiation and carbozincation reactions are typically only observed on activated or strained alkenes and alkynes.[10] For example, electron withdrawing groups like esters, nitriles or sulfones must be in conjugation with the carbon-carbon π-system (see Michael reaction) or a directing group like an alcohol or amine must be nearby to direct the reaction. These reactions can be catalyzed by a variety of transition metals such as iron,[10][11] copper,[10] zirconium,[12] nickel,[10][13] cobalt[14] and others.
Illustrative is the Fe-catalyzed reaction of methylphenylacetylene with phenylmagnesium bromide, which generates a vinyl magnesium intermediate. Hydrolysis affords the diphenylalkene:[11][15]
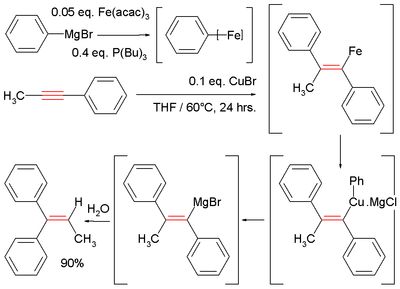
Arylmagnesiation

Carbopalladation
[edit]Carbopalladations can be a description of the elementary step of a reaction catalyzed by a palladium catalyst (Mizoroki-Heck reaction)[16] and can also refer to a carbometalation reaction with a palladium catalyst (alkene difunctionalization,[17] hydrofunctionalization,[18][19] or reductive Heck[20])
References
[edit]- ^ a b Negishi, Ei-ichi; Tan, Ze (2005), "Diastereoselective, Enantioselective, and Regioselective Carboalumination Reactions Catalyzed by Zirconocene Derivatives", Metallocenes in Regio- and Stereoselective Synthesis: -/-, Topics in Organometallic Chemistry, Springer Berlin Heidelberg, pp. 139–176, doi:10.1007/b96003, ISBN 9783540314523
- ^ a b Xu, Shiqing; Negishi, Ei-ichi (2016-10-18). "Zirconium-Catalyzed Asymmetric Carboalumination of Unactivated Terminal Alkenes". Accounts of Chemical Research. 49 (10): 2158–2168. doi:10.1021/acs.accounts.6b00338. ISSN 0001-4842. PMID 27685327.
- ^ Ma, Shengming; Negishi, Ei-ichi (1997-02-01). "Anti-Carbometalation of Homopropargyl Alcohols and Their Higher Homologues via Non-Chelation-Controlled Syn-Carbometalation and Chelation-Controlled Isomerization". The Journal of Organic Chemistry. 62 (4): 784–785. doi:10.1021/jo9622688. ISSN 0022-3263.
- ^ a b c O’Shea, Donal F.; Hogan, Anne-Marie L. (2008-08-18). "Synthetic applications of carbolithiation transformations". Chemical Communications (33): 3839–3851. doi:10.1039/B805595E. ISSN 1364-548X. PMID 18726011.
- ^ García, Graciela V.; Nudelman, Norma Sbarbati (2009-02-11). "Tandem Reactions Involving Organolithium Reagents. A Review". Organic Preparations and Procedures International. 35 (5): 445–500. doi:10.1080/00304940309355860. S2CID 98358106.
- ^ Norsikian, Stephanie; Marek, Ilane; Normant, Jean-F (1997-10-27). "Enantioselective Carbolithiation of β-Alkylated Styrene". Tetrahedron Letters. 38 (43): 7523–7526. doi:10.1016/S0040-4039(97)10022-3. ISSN 0040-4039.
- ^ Norsikian, Stephanie; Marek, Ilan; Klein, Sophie; Poisson, Jean F.; Normant, Jean F. (1999). "Enantioselective Carbometalation of Cinnamyl Derivatives: New Access to Chiral Disubstituted Cyclopropanes— Configurational Stability of Benzylic Organozinc Halides". Chemistry – A European Journal. 5 (7): 2055–2068. doi:10.1002/(SICI)1521-3765(19990702)5:7<2055::AID-CHEM2055>3.0.CO;2-9. ISSN 1521-3765.
- ^ Tait, Michael; Donnard, Morgan; Minassi, Alberto; Lefranc, Julien; Bechi, Beatrice; Carbone, Giorgio; O'Brien, Peter; Clayden, Jonathan (2013). "Amines Bearing Tertiary Substituents by Tandem Enantioselective Carbolithiation-Rearrangement of Vinylureas". Organic Letters. 15 (1): 34–37. doi:10.1021/ol3029324. ISSN 1523-7060. PMID 23252812.
- ^ McKinley, Neola F.; O'Shea, Donal F. (2006). "Carbolithiation of Diphenylacetylene as a Stereoselective Route to (Z)-Tamoxifen and Related Tetrasubstituted Olefins". J. Org. Chem. (Note). 71 (25): 9552–9555. doi:10.1021/jo061949s. PMID 17137396.
- ^ a b c d Yorimitsu, Hideki; Murakami, Kei (2013-02-11). "Recent advances in transition-metal-catalyzed intermolecular carbomagnesiation and carbozincation". Beilstein Journal of Organic Chemistry. 9 (1): 278–302. doi:10.3762/bjoc.9.34. ISSN 1860-5397. PMC 3596116. PMID 23503106.
- ^ a b Shirakawa, Eiji; Yamagami, Takafumi; Kimura, Takahiro; Yamaguchi, Shigeru; Hayashi, Tamio (2005). "Arylmagnesiation of Alkynes Catalyzed Cooperatively by Iron and Copper Complexes". J. Am. Chem. Soc. (Communication). 127 (49): 17164–17165. doi:10.1021/ja0542136. PMID 16332046.
- ^ Negishi, Eiichi; Miller, Joseph A. (1983-10-01). "Selective carbon-carbon bond formation via transition metal catalysis 37. Controlled carbometalation. 16. Novel syntheses of .alpha.,.beta.-unsaturated cyclopentenones via allylzincation of alkynes". Journal of the American Chemical Society. 105 (22): 6761–6763. doi:10.1021/ja00360a060. ISSN 0002-7863.
- ^ Xue, Fei; Zhao, Jin; Hor, T. S. Andy; Hayashi, Tamio (2015-03-11). "Nickel-Catalyzed Three-Component Domino Reactions of Aryl Grignard Reagents, Alkynes, and Aryl Halides Producing Tetrasubstituted Alkenes". Journal of the American Chemical Society. 137 (9): 3189–3192. doi:10.1021/ja513166w. ISSN 0002-7863. PMID 25714497.
- ^ Murakami, Kei; Yorimitsu, Hideki; Oshima, Koichiro (2010). "Cobalt-Catalyzed Benzylzincation of Alkynes". Chemistry – A European Journal. 16 (26): 7688–7691. doi:10.1002/chem.201001061. ISSN 1521-3765. PMID 20521290.
- ^ In this reaction the Grignard reagent combines with iron acetylacetonate and tributylphosphine to give an ill-defined aryliron intermediate, which then reacts with copper(I) chloride an intermediate cuprate.
- ^ Negishi, Ei-ichi; Copéret, Christophe; Ma, Shengming; Liou, Show-Yee; Liu, Fang (January 1996). "Cyclic Carbopalladation. A Versatile Synthetic Methodology for the Construction of Cyclic Organic Compounds". Chemical Reviews. 96 (1): 365–394. doi:10.1021/cr950020x. ISSN 0009-2665. PMID 11848757.
- ^ Sigman, Matthew S.; Jensen, Katrina H. (2008-10-30). "Mechanistic approaches to palladium-catalyzed alkene difunctionalization reactions". Organic & Biomolecular Chemistry. 6 (22): 4083–4088. doi:10.1039/B813246A. ISSN 1477-0539. PMC 2656348. PMID 18972034.
- ^ Engle, Keary M.; McAlpine, Indrawan; Marsters, Rohan P.; Wang, Fen; He, Mingying; Yang, Shouliang; Gallego, Gary M.; Yang, Kin S.; Hill, David E. (2018-11-14). "Palladium(II)-catalyzed γ-selective hydroarylation of alkenyl carbonyl compounds with arylboronic acids". Chemical Science. 9 (44): 8363–8368. doi:10.1039/C8SC03081B. ISSN 2041-6539. PMC 6247822. PMID 30542583.
- ^ O’Duill, Miriam L.; Matsuura, Rei; Wang, Yanyan; Turnbull, Joshua L.; Gurak, John A.; Gao, De-Wei; Lu, Gang; Liu, Peng; Engle, Keary M. (2017-11-08). "Tridentate Directing Groups Stabilize 6-Membered Palladacycles in Catalytic Alkene Hydrofunctionalization". Journal of the American Chemical Society. 139 (44): 15576–15579. doi:10.1021/jacs.7b08383. ISSN 0002-7863. PMC 6002750. PMID 28972751.
- ^ Gurak, John A.; Engle, Keary M. (2018-10-05). "Practical Intermolecular Hydroarylation of Diverse Alkenes via Reductive Heck Coupling". ACS Catalysis. 8 (10): 8987–8992. doi:10.1021/acscatal.8b02717. PMC 6207086. PMID 30393575.