
Carbonyl oxidation with hypervalent iodine reagents
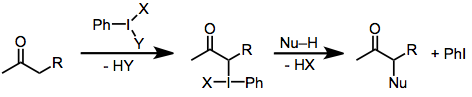
Carbonyl oxidation with hypervalent iodine reagents involves the functionalization of the α position of carbonyl compounds through the intermediacy of a hypervalent iodine(III) enolate species. This electrophilic intermediate may be attacked by a variety of nucleophiles or undergo rearrangement or elimination.[1]
Introduction[edit]
Hypervalent iodine(III) compounds are attractive oxidizing agents because of their stability and selectivity. In the presence of enolizable carbonyl compounds, they are able to accomplish oxidative functionalization of the α position. A key iodine(III) enolate intermediate forms, which then undergoes either nucleophilic substitution (α-functionalization), elimination (dehydrogenation), or rearrangement. Common hypervalent iodine reagents used to effect these transformations include iodosylbenzene (PhIO),[2] iodobenzene diacetate (PhI(OAc)2),[3] Koser's reagent (PhI(OTs)OH),[4] and (dichloroiodo)benzene (PhICl2).[5]
-
 (1)
(1)

Mechanism and stereochemistry[edit]
Prevailing mechanism[edit]
The mechanism of carbonyl oxidation by iodine(III) reagents varies as a function of substrate structure and reaction conditions, but some generalizations are possible. Under basic conditions, the active iodinating species are iodine(III) compounds in which any relatively acidic ligands on iodine (such as acetate) have been replaced by alkoxide.[2] In all cases, the α carbon forms a bond to iodine. Reduction of iodine(III) to iodine(I) then occurs via attack of a nucleophile on the now electrophilic α carbon. Under basic conditions, nucleophilic attack at the carbonyl carbon is faster than attack at the α carbon. Iodine displacement is actually accomplished intramolecularly by the carbonyl oxygen, which becomes the α-hydroxyl oxygen in the product.[6]
-
 (2)
(2)

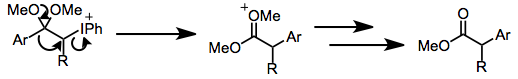
Rearrangements of the iodine(III) enolate species have been observed. Under acidic conditions, oxidations of aryl enol ethers lead to α-aryl esters via 1,2-aryl migration.[7] Ring-contractive Favorskii rearrangements may take place under basic conditions (see equation (12) below).
-
 (3)
(3)

Stereochemistry[edit]
Using a chromium carbonyl complex, it was shown that displacement of iodine likely occurs with inversion of configuration. Iodine approaches on the side opposite the chromium tricarbonyl unit due to steric hindrance. Invertive displacement leads to a syn relationship between chromium and the α hydroxyl group.[8]
-
 (4)
(4)

Studies on the oxidation of unsaturated carbonyl compounds also provide stereochemical insight. Only the isomer with a syn relationship between the α-hydroxy and β-methoxy groups was observed. After nucleophilic attack by methoxide, iodine approaches the face opposite the methoxide. Invertive displacement by hydroxide then leads to the syn isomer.[9]
-
 (5)
(5)

Scope and limitations[edit]
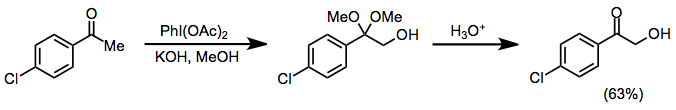
Under protic conditions, ketones undergo α-hydroxylation and dimethyl acetal formation. Both iodosylbenzene and iodobenzene diacetate (IBD) can effect this transformation. This method can be used to synthesize α-hydroxy ketones after acidic hydrolysis of the ketal functionality.[10]
-
 (6)
(6)

In the presence of diaryliodonium salts, enolates undergo α-arylation. Bulky diaryliodoniums react more slowly, and enolate homocoupling (see equation (10) below) begins to compete as the aromatic ring is substituted.[11]
-
 (7)
(7)

α-Oxytosylation facilitates the elaboration of carbonyl compounds into a variety of α-functionalized products. The α-tosyloxycarbonyl compounds that result are more stable than α-halocarbonyl compounds and are not lachrymators.[12]
-
 (8)
(8)

Silyl enol ethers undergo many of the same reactions as carbonyl compounds in the presence of iodine(III) reagents. α-Alkoxylation is possible in the presence of an external alcohol nucleophile, although yields are somewhat variable.[13]
-
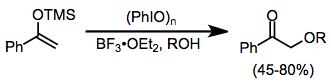 (9)
(9)

When no external or internal nucleophile is present, oxidative homocoupling occurs, yielding 1,4-dicarbonyl compounds.[14]
-
 (10)
(10)

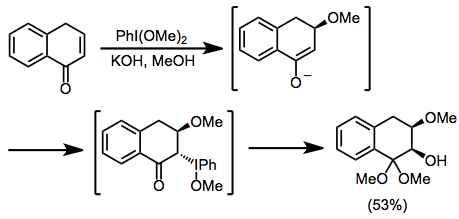
Intramolecularly tethered nucleophiles may displace iodobenzene to afford lactones or other heterocycles.[15] If acidic hydrogens are present in the cyclic product, overoxidation can occur under the reaction conditions.
-
 (11)
(11)

In some cases, rearrangements complicate hypervalent iodine oxidations of carbonyl compounds. Aryl migration may occur under acidic conditions, yielding α-aryl esters from enol ethers.[7] Favorskii rearrangements have also been observed, and these have been particularly useful for steroid synthesis.[16]
-
 (12)
(12)

Synthetic applications[edit]
Oxidative functionalization of silyl enol ethers in low concentration (to avoid homocoupling) without an external nucleophile leads to dehydrogenation. This can be a useful way to generate α,β-unsaturated carbonyl compounds in the absence of functional handles. For instance, dehydrogenation is employed in steroid synthesis to form unsaturated ketones.[17]
-
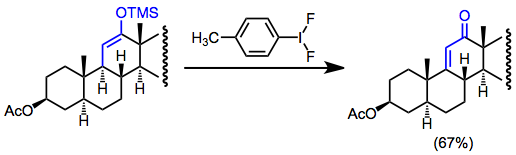 (13)
(13)

Comparison with other methods[edit]
Few compounds that oxidize carbonyl compounds rival the safety, selectivity, and versatility of hypervalent iodine reagents. Other methods for the α-hydroxylation of carbonyl compounds may employ toxic organometallic compounds (such as lead tetraacetate or osmium tetroxide). One alternative to hypervalent iodine oxidation that does not employ heavy metals is the attack of a metal enolate on dioxygen, followed by reduction of the resulting peroxide (equation (14)). The most popular method for the α-hydroxylation of carbonyl compounds is the Rubottom oxidation, which employs silyl enol ethers as substrates and peracids as oxidants.[18]
-
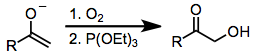 (14)
(14)

Oxidative rearrangements are generally easier to accomplish using hypervalent iodine reagents than other oxidizing agents. The Willgerodt-Kindler reaction of alkyl aryl ketones, for instance, requires forcing conditions and often gives low yields of amide products.
-
 (15)
(15)

Experimental conditions and procedure[edit]
Example Procedure[edit]
-
 (16)
(16)

Hydroxy(mesyloxy)iodobenzene (3.16 g, 10 mmol) was added to a solution of the methyl trimethylsilyl phenylketene acetal derived from methyl phenylacetate (3.33 g, 15 mmol) in dry dichloromethane (50 mL). The mixture was stirred at room temperature for 2 hours and then washed with aqueous sodium bicarbonate solution (3 × 50 mL). The organic phase was dried (MgSO4) and concentrated in vacuo to yield the crude mesyloxyester which was purified by column chromatography on silica gel (hexane-dichloromethane, 1:1) to give 1.58 g (65%) of the title compound, mp 91–92°; IR (KBr) 1760 cm−1 (CO); 1H NMR (CDCl3): δ 3.10 (s, 3H), 3.80 (s, 3H), 6.00 (s, H), 7.40–7.80 (m, 5H); 13C NMR (CDCl3): δ 168.2 (s), 132.2 (s), 130.0 (s), 129.0 (s), 127.7 (s), 78.9 (s), 53.0 (s), 39.45 (s); MS, m/z 185 (53), 165 (15), 145 (15), 107 (100), 90 (12), 79 (65), 51 (17). [19]
References[edit]
- ^ Moriarty, R. M.; Prakash, O. Org. React. 1999, 54, 273. doi:10.1002/0471264180.or054.02
- ^ a b Schardt, B. C.; Hill, C. L. Inorg. Chem. 1983, 22, 1563.
- ^ Moriarty, R. M.; Hu, H. Tetrahedron Lett. 1981, 22, 2747.
- ^ Koser, G. F.; Relenyi, A. G.; Kalos, A. N.; Rebrovic, L.; Wettach, R. H. J. Org. Chem. 1982, 47, 2487.
- ^ Dneprovskii, A. S.; Krainyuchenko, I. V.; Temnikova, T. I. J. Org. Chem. USSR (Engl. Transl.) 1978, 14, 1414.
- ^ Moriarty, R. M.; Hu, H.; Gupta, S. C. Tetrahedron Lett. 1981, 22, 1283.
- ^ a b Prakash, O. Aldrichimica Acta 1995, 28, 63.
- ^ Moriarty, R. M.; Engerer, S. C.; Prakash, O.; Prakash, I.; Gill., U. S.; Freeman, W. A. J. Org. Chem. 1987, 52, 153.
- ^ Tamura, Y.; Yakura, T.; Terashi, H.; Haruta, J.; Kita, Y. Chem. Pharm. Bull. 1987, 35, 570.
- ^ Podolesov, B. J. Org. Chem. 1984, 49, 2644.
- ^ Beringer, F. M.; Galton, S. A. J. Org. Chem. 1963, 28, 3417.
- ^ Prakash, O.; Goyal, S. Synthesis 1992, 6291.
- ^ Moriarty, R. M.; Prakash, O.; Duncan, M. P.; Vaid, R. K.; Musallam, H. A. J. Org. Chem. 1987, 52, 150.
- ^ Moriarty, R. M.; Prakash, O.; Duncan, M. P. J. Chem. Soc., Chem. Commun. 1985, 420.
- ^ Moriarty, R. M.; Prakash, O.; Prakash, I.; Musallam, H. A. J. Chem. Soc., Chem. Commun. 1984, 1342.
- ^ Daum, S. J. Tetrahedron Lett. 1984, 25, 4725.
- ^ Tsushima, T.; Kawada, K.; Tsuji, T. Tetrahedron Lett. 1982, 23, 1165.
- ^ Chen, B.-C.; Zhou, P.; Davis, F. A.; Ciganek, E. Org. React. 2003, 62, 1.
- ^ Moriarty, R. M.; Penmasta, R.; Awasthi, A. K.; Epa, R. W.; Prakash, I. J. Org. Chem. 1989, 54, 1101.