
User:Smokefoot/sandbox2
Metal borohydride complexes[edit]
Ce(BH4)3 made by ball-milling[1]
Li[Zn2(BH4)5] and M[Zn(BH4)3] salts (M=Na, K)[2]
Zr(BH4)4[3]
Ti(BH4)3(OEt2)Cite error: A <ref> tag is missing the closing </ref> (see the help page).
Chemical place names[edit]
Antimony, U Barium Springs, NC Boron, CA Bromide, OK Carbon, IN, TX, WV Chloride, AZ, MO, NM Cobalt, CO Iron, MN Krypton, KY Lithium, MO Mercury, TX Neon, KY Platinum, AK Radium, CO, KS, MN, AS Silver, AS Sulfur, IN, KY, LA, NV, OK, SD Tungsten, CO Vanadium, NM Zinc, AR
Asymmetric hydrogenation is a chemical reaction that adds two atoms of hydrogen preferentially to one of two faces of an unsaturated substrate molecule, such as an alkene or ketone. The selectivity derives from the manner that the substrate binds to the chiral catalysts. In jargon, this binding transmits spatial information (what chemists refer to as chirality) from the catalyst to the target, favoring the product as a single enantiomer. This "enzyme-like selectivity" is applied to the synthesis of some commercialpharmaceutical agents and agrochemicals.

History[edit]
In 1956 a heterogeneous catalyst made of palladium deposited on silk was shown to effect asymmetric hydrogenation.**[4] Later, in 1968, the groups of William Knowles and Leopold Horner independently published the examples of asymmetric hydrogenation using a homogeneous catalysts. While exhibiting only modest enantiomeric excesses, these early reactions demonstrated feasibility. By 1972, enantiomeric excess of 90% was achieved, and the first industrial synthesis of the Parkinson's drug L-DOPA commenced using this technology.[5][6]
.svg)
The field of asymmetric hydrogenation continued to experience a number of notable advances. Henri Kagan developed DIOP, an easily prepared C2-symmetric diphosphine that gave high ee's in certain reactions. Ryōji Noyori introduced the ruthenium-based catalysts for the asymmetric hydrogenated polar substrates, such as ketones and aldehydes. The introduction of P,N ligands then further expanded the scope of the C2-symmetric ligands, although they are not fundamentally superior to chiral ligands lacking rotational symmetry.[7] Today, asymmetric hydrogenation is a routine methodology in laboratory and industrial scale organic chemistry.
The importance of asymmetric hydrogenation was recognized by the 2001 Nobel Prize in Chemistry awarded to William Standish Knowles and Ryōji Noyori.
Mechanism[edit]
Inner sphere mechanisms[edit]
Two major mechanisms have been proposed for catalytic hydrogenation with rhodium complexes: the unsaturated mechanism and the dihydride mechanism. While distinguishing between the two mechanisms is difficult, the difference between the two for asymmetric hydrogenation is relatively unimportant since both converge to a common intermediate before any stereochemical information is transferred to the product molecule.**[8]

The preference for producing one enantiomer instead of another in these reactions is often explained in terms of steric interactions between the ligand and the prochiral substrate. Consideration of these interactions has led to the development of quadrant diagrams where "blocked" areas are denoted with a shaded box, while "open" areas are left unfilled. In the modeled reaction, large groups on an incoming olefin will tend to orient to fill the open areas of the diagram, while smaller groups will be directed to the blocked areas and hydrogen delivery will then occur to the back face of the olefin, fixing the stereochemistry. Note that only part of the chiral phosphine ligand is shown for the sake of clarity.

Metals[edit]
Platinum-group metals[edit]
Rhodium, the first metal to be used in a homogeneous asymmetric hydrogenation,**[9] continues to be widely used. Targets for asymmetric hydrogenation with rhodium generally require a coordinating group close to the olefin.[8] While this requirement is a limitation, many classes of substrates possess such functionalization, e.g. unsaturated amides.**[10]
The Noyori asymmetric hydrogenation is based on ruthenium. Subsequent work has expanded upon Noyori's original catalyst template, leading to the inclusion of traditionally difficult substrates like t-butyl ketones as viable substrates for hydrogenation with ruthenium catalysts. Transfer hydrogenation based on the Ru and TsDPEN has also enjoyed commercial success.**[11]
Iridium catalysts are useful for a number of "non-traditional" substrates for which good catalysts had not been found with Ru and Rh.**[12] Unfunctionalized olefins**[13] exist. A common difficulty with iridium-based catalyst is their tendency to trimerize in solution.[13] The use of the non-coordinating anion BArF
4− has proven to be the most widely applicable solution to the aggregation problem.[13]**[14] Other strategies to enhance catalyst stability include the addition of an additional coordinating arm to the chiral ligand,[15] increasing the steric bulk of the ligand, using a dendrimeric ligand, increasing the rigidity of the ligand, immobilizing the ligand, and using heterobimetallic systems (with iridium as one of the metals).[16]
Base metals[edit]
Iron is a popular research target for many catalytic processes, owing largely to its low cost and low toxicity relative to other transition metals.**[17] Asymmetric hydrogenation methods using iron have been realized, although in terms of rates and selectivity, they are inferior to catalysts based on precious metals.
Ligand classes[edit]
Phosphine ligands[edit]
Chiral phosphine ligands, especially C2-symmetric ligands, are the source of chirality in most asymmetric hydrogenation catalysts. Of these the BINAP ligand is well-known, as a result of its Nobel Prize-winning application in the Noyori asymmetric hydrogenation.[5]
Chiral phosphine ligands can be generally classified as mono- or bidentate. They can be further classified according to the location of the stereogenic centre – phosphorus vs the organic substituents. Ligands with a C2 symmetry element have been particularly popular, in part because the presence of such an element reduces the possible binding conformations of a substrate to a metal-ligand complex dramatically (often resulting in exceptional enantioselectivity).**[18]
Monodentate phosphines[edit]
Monophosphine-type ligands were among the first to appear in asymmetric hydrogenation, e.g., the ligand CAMP.[19] Continued research into these types of ligands has explored both P-alkyl and P-heteroatom bonded ligands, with P-heteroatom ligands like the phosphites and phosphoramidites generally achieving more impressive results.**[20] Structural classes of ligands that have been successful include those based on the binapthyl structure of MonoPHOS or the spiro ring system of SiPHOS. Notably, these monodentate ligands can be used in combination with each other to achieve a synergistic improvement in enantioselectivity; something that is not possible with the diphosphine ligands.[20]



Chiral diphosphine ligands[edit]
The diphosphine ligands have received considerably more attention than the monophosphines and, perhaps as a consequence, have a much longer list of achievement. This class includes the first ligand to achieve high selectivity (DIOP), the first ligand to be used in industrial asymmetric synthesis (DIPAMP[21][22]**[6]) and what is likely the best known chiral ligand (BINAP).[5] Chiral diphosphine ligands are now ubiquitous in asymmetric hydrogenation.

P,N and P,O ligands[edit]


The use of P,N ligands in asymmetric hydrogenation can be traced to the C2 symmetric bisoxazoline ligand.[23] However, these symmetric ligands were soon superseded by monooxazoline ligands whose lack of C2 symmetry has in no way limits their efficacy in asymmetric catalysis.**[24] Such ligands generally consist of an achiral nitrogen-containing heterocycle that is functionalized with a pendant phosphorus-containing arm, although both the exact nature of the heterocycle and the chemical environment phosphorus center has varied widely. No single structure has emerged as consistently effective with a broad range of substrates, although certain privileged structures (like the phosphine-oxazoline or PHOX architecture) have been established.[25] Moreover, within a narrowly defined substrate class the performance of metallic complexes with chiral P,N ligands can closely approach perfect conversion and selectivity in systems otherwise very difficult to target. Certain complexes derived from chelating P-O ligands have shown promising results in the hydrogenation of α,β-unsaturated ketones and esters.
Acyclic substrates[edit]
Acyclic unsaturated substrates (olefins, ketones, enamines imines) represents the most common prochiral substrates. Substrates that are particularly amenable to asymmetric hydrogenation often feature a polar functional group adjacent to the site to be hydrogenated. In the absence of this functional group, catalysis often results in low ee's. For unfunctionalized olefins, iridium with P,N-based ligands) have proven successful catalysts. Catalyst utility within this category is unusually narrow; consequently, many different categories of solved and unsolved catalytic problems have developed. 1,1-disubstituted, 1,2-diaryl trisubstituted, 1,1,2-trialkyl and tetrasubstituted olefins represent classes that have been investigated separately, and even within these classes variations may exist that make different solutions optimal.


Conversely to the case of olefins, asymmetric hydrogenation of enamines has favoured diphosphine-type ligands; excellent results have been achieved with both iridium- and rhodium-based systems. However, even the best systems often suffer from low ee's and a lack of generality. Certain pyrrolidine-derived enamines of aromatic ketones are amenable to asymmetrically hydrogenation with cationic rhodium(I) phosphonite systems, and I2 and acetic acid system with ee values usually above 90% and potentially as high as 99.9%. A similar system using iridium(I) and a very closely related phosphoramidite ligand is effective for the asymmetric hydrogenation of pyrrolidine-type enamines where the double bond was inside the ring: in other words, of dihydropyrroles.
Imines and ketones[edit]

Ketones and imines are related functional groups, and effective technologies for the asymmetric hydrogenation of each are also closely related. Of these, Noyori's ruthenium-chiral diphosphine-diamine system is perhaps one of the best known. It can be employed in conjunction with a wide range of phosphines and amines (where the amine may be, but need not be, chiral) and can be easily adjusted for an optimal match with the target substrate, generally achieving enantiomeric excesses (ee's) above 90%.**[26]**[27]
Iridium/P,N ligand-based systems are also commonly used for the asymmetric hydrogenation of ketones and imines. For example, a consistent system for benzylic aryl imines uses the P,N ligand SIPHOX in conjunction with iridium(I) in a cationic complex to achieve asymmetric hydrogenation with ee >90%.[28] One of the most efficient and effective catalysts ever developed for the asymmetric hydrogenation of ketones, with a turnover number (TON) up to 4,550,000 and ee up to 99.9%, uses another iridium(I) system with a closely related tridentate ligand.[15]

Aromatic substrates[edit]
The asymmetric hydrogenation of aromatic (especially heteroaromatic), substrates is a very active field of ongoing research. Catalysts in this field must contend with a number of complicating factors, including the tendency of highly stable aromatic compounds to resist hydrogenation, the potential coordinating (and therefore catalyst-poisoning) abilities of both substrate and product, and the great diversity in substitution patterns that may be present on any one aromatic ring.**[29] Of these substrates the most consistent success has been seen with nitrogen-containing heterocycles, where the aromatic ring is often activated either by protonation or by further functionalization of the nitrogen (generally with an electron-withdrawing protecting group). Such strategies are less applicable to oxygen- and sulfur-containing heterocycles, since they are both less basic and less nucleophilic; this additional difficulty may help to explain why few effective methods exist for their asymmetric hydrogenation.
Quinolines, isoquinolines and quinoxalines[edit]
Two systems exist for the asymmetric hydrogenation of 2-substituted quinolines with isolated yields generally greater than 80% and ee values generally greater than 90%. While the first chiral phosphine used in this system was MeOBiPhep, newer iterations have focused on improving the performance of this ligand. To this end, systems use phosphines (or related ligands) with improved air stability, recyclability, ease of preparation,[30] lower catalyst loading and the potential role of achiral phosphine additives. As of October 2012 no mechanism appears to have been proposed, although both the necessity of I2 or a halogen surrogate and the possible role of the heteroaromatic N in assisting reactivity have been documented.[29]

Pyridines[edit]
Pyridines are highly variable substrates for asymmetric reduction (even compared to other heteroaromatics), in that five carbon centers are available for differential substitution on the initial ring. As of October 2012 no method seems to exist that can control all five, although at least one reasonably general method exists.
The most-general method of asymmetric pyridine hydrogenation is actually a heterogeneous method, where asymmetry is generated from a chiral oxazolidinone bound to the C2 position of the pyridine. Hydrogenating such functionalized pyridines over a number of different heterogeneous metal catalysts gave the corresponding piperidine with the substituents at C3, C4, and C5 positions in an all-cis geometry, in high yield and excellent enantioselectivity. The oxazolidinone auxiliary is also conveniently cleaved under the hydrogenation conditions.

Methods designed specifically for 2-substituted pyridine hydrogenation can involve asymmetric systems developed for related substrates like 2-substituted quinolines and quinoxalines. For example, an iridium(I)\chiral phosphine\I2 system is effective in the asymmetric hydrogenation of activated (alkylated) 2-pyridiniums.[31]
Indoles[edit]
The asymmetric hydrogenation of indoles initially focused on N-protected indoles, where the protecting group could serve both to activate the heterocycle to hydrogenation and as a secondary coordination site for the metal. Later work allowed unprotected indoles to be targeted through Brønsted acid activation of the indole.
In the initial report on asymmetric indole hydrogenation, N-acetyl 2-substituted indoles could be protected with high yields and ee of 87-95%. 3-substituted indoles were less successful, with hydrolysis of the protecting group outcompeting the hydrogenation of the indole.

Despite these advances in the asymmetric hydrogenation of protected indoles, considerable operational simplicity can be gained by removing the protecting group altogether. This has been achieved with catalytic systems utilizing Brønsted acids to activate the indole. The initial system used a Pd(TFA)2/H8-BINAP system to achieve the enantioselective cis-hydrogenation of 2,3- and 2-substituted indoles with high yield and excellent ee. A similar process, where sequential Friedel-Crafts alkylation and asymmetric hydrogenation occur in one pot, allow asymmetric 2,3-substituted indolines to be selectively prepared from 2-substituted indoles in similarly high yields and ee.

A promising organocatalytic method for the asymmetric hydrogenation of 2,3-substituted indoles utilizing a chiral Lewis base also exists, although the observed ee's are not quite equivalent to those of the metal-based hydrogenations.[32]
Pyrroles[edit]
Achieving complete conversion of pyrroles to pyrrolidines by asymmetric hydrogenation has so far proven difficult, with partial-hydrogenation products often being observed.[33][34] Complete enantioselective reduction is possible, with the outcome depending on both the starting substrate and the method.
The asymmetric hydrogenation of 2,3,5-substituted pyrroles was achieved by the recognition that such substrates bear the same substitution pattern as 2-substituted indoles, and an asymmetric hydrogenation system that is effective for one of these substrates might be effective for both. Such an analysis led to the development of a ruthenium(I)/phosphine/amine base system for 2,3,5-substituted N-Boc pyrroles that can give either dihydro or tetrahydropyrroles (pyrrolidines), depending on the nature of the pyrrole substituents. An all-phenyl substitution pattern leads to dihydropyrroles in very high yield (>96%) and essentially perfect enantioselectivity. Access to the fully hydrogenated, all-cis dihydropyrrole may then be accessible through diastereoselective heterogeneous hydrogenation. Alkyl substitution may lead to either the dihydro or tetrahydropyrrole, although the yields (>70%) and enantioselectivities (often >90%) generally remain high. The regioselectivity in both cases appears to be governed by sterics, with the less-substituted double being preferentially hydrogenated.[33]

Unprotected 2,5-pyrroles may also be hydrogenated asymmetrically by a Brønsted acid/Pd(II)/chiral phosphine-catalyzed method, to give the corresponding 2,5-disubstituted 1-pyrrolines in roughly 70-80% yield and 80-90% ee.[34]
Oxygen-containing heterocycles[edit]
The asymmetric hydrogenation of furans and benzofurans has so far proven challenging.**[35] Some Ru-NHC complex catalyze asymmetric hydrogenations of benzofurans.

Sulfur-containing heterocycles[edit]
As is the case with oxygen-containing heterocycles, the asymmetric hydrogenation of compounds where sulfur is part of the initial unsaturated pi-bonding system so far appears to be limited to thiophenes and benzothiophenes. The key approach to the asymmetric hydrogenation of these heterocycles involves a ruthenium(II) catalyst and chiral, C2 symmetric N-heterocyclic carbene (NHC). This system appears to possess superb selectivity (ee > 90%) and perfect diastereoselectivity (all cis) if the substrate has a fused (or directly bound) phenyl ring but yields only racemic product in all other tested cases.[36]

Heterogeneous catalysis[edit]
No heterogeneous catalyst has been commercialized for asymmetric hydrogenation.
The first asymmetric hydrogenation focused on palladium deposited on a silk support. Cinchona alkaloids have been used as chiral modifiers for enantioselectivity hydrogenation.**[37]

== Industrial applications ==

Knowles' research into asymmetric hydrogenation and its application to the production scale synthesis of L-Dopa[6] gave asymmetric hydrogenation a strong start in the industrial world. A 2001 review indicated that asymmetric hydrogenation accounted for 50% of production scale, 90% of pilot scale, and 74% of bench scale catalytic, enantioselective processes in industry, with the caveat that asymmetric catalytic methods in general were not yet widely used.**[38]
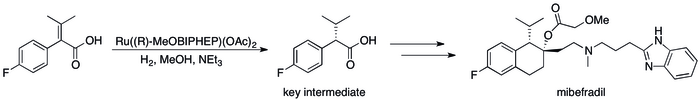
The success of asymmetric hydrogenation in industry**[39] can be seen in a number of specific cases where the replacement of kinetic resolution based methods has resulted in substantial improvements in the process's efficiency. For example, Roche's Catalysis Group was able to achieve the synthesis of (S,S)-Ro 67-8867 in 53% overall yield, a dramatic increase above the 3.5% that was achieved in the resolution based synthesis.**[40] Roche's synthesis of mibefradil was likewise improved by replacing resolution with asymmetric hydrogenation, reducing the step count by three and increasing the yield of a key intermediate to 80% from the original 70%.[41]
References[edit]
- ^ Gennari, F.C.; Esquivel, M.R. (2009). "Synthesis and dehydriding process of crystalline Ce(BH4)3". Journal of Alloys and Compounds. 485 (1–2): L47–L51. doi:10.1016/j.jallcom.2009.06.078.
- ^ Jaroń, Tomasz; Orłowski, Piotr A.; Wegner, Wojciech; Fijałkowski, Karol J.; Leszczyński, Piotr J.; Grochala, Wojciech (2015). "Hydrogen Storage Materials: Room‐Temperature Wet‐Chemistry Approach toward Mixed‐Metal Borohydrides". Angewandte Chemie International Edition. 54 (4): 1236–1239. doi:10.1002/anie.201408456. PMID 25470241.
- ^ 12-coordinate?.. doi:/10.1039/C19670000403.
{{cite journal}}: Check|doi=value (help); Cite journal requires|journal=(help); Missing or empty|title=(help) - ^ Akabori, S.; Sakurai, S.; Izumi, Y.; Fujii, Y. (1956). "An Asymmetric Catalyst". Nature. 178 (4528): 323. Bibcode:1956Natur.178..323A. doi:10.1038/178323b0. PMID 13358737. S2CID 4221816.
- ^ a b c Noyori, R. (2003). "Asymmetric Catalysis: Science and Opportunities (Nobel Lecture 2001)". Advanced Synthesis & Catalysis. 345 (12): 15–41. doi:10.1002/adsc.200390002.
- ^ a b c Knowles, W. S. (2002). "Asymmetric Hydrogenations (Nobel Lecture)". Angewandte Chemie International Edition. 41 (12): 1998–2007. doi:10.1002/1521-3773(20020617)41:12<1998::AID-ANIE1998>3.0.CO;2-8. PMID 19746594.
- ^ Pfaltz, A. (2004). "Asymmetric Catalysis Special Feature Part II: Design of chiral ligands for asymmetric catalysis: From C2-symmetric P,P- and N,N-ligands to sterically and electronically nonsymmetrical P,N-ligands". Proceedings of the National Academy of Sciences. 101 (16): 5723–5726. Bibcode:2004PNAS..101.5723P. doi:10.1073/pnas.0307152101. PMC 395974. PMID 15069193.
- ^ a b Gridnev, I. D.; Imamoto, T. (2004). "On the Mechanism of Stereoselection in Rh-Catalyzed Asymmetric Hydrogenation: A General Approach for Predicting the Sense of Enantioselectivity". Accounts of Chemical Research. 37 (9): 633–644. doi:10.1021/ar030156e. PMID 15379579.
- ^ Knowles, W. S.; Sabacky, M. J. (1968). "Catalytic asymmetric hydrogenation employing a soluble, optically active, rhodium complex". Chemical Communications (London) (22): 1445. doi:10.1039/C19680001445.
- ^ Pilkington, C.; Lennon, I. (2003). "The Application of Asymmetric Hydrogenation for the Manufacture of Pharmaceutical Intermediates:The Need for Catalyst Diversity". Synthesis. 2003 (11): 1639. doi:10.1055/s-2003-40871.
- ^ Ikariya, T.; Blacker, A. J. (2007). "Asymmetric Transfer Hydrogenation of Ketones with Bifunctional Transition Metal-Based Molecular Catalysts". Accounts of Chemical Research. 40 (12): 1300–1308. doi:10.1021/ar700134q. PMID 17960897.
- ^ Church, T. L.; Andersson, P. G. (2008). "Iridium catalysts for the asymmetric hydrogenation of olefins with nontraditional functional substituents". Coordination Chemistry Reviews. 252 (5–7): 513. doi:10.1016/j.ccr.2007.09.015.
- ^ a b c Pfaltz, A.; Blankenstein, J. R.; Hilgraf, R.; Hörmann, E.; McIntyre, S.; Menges, F.; Schönleber, M.; Smidt, S. P.; Wüstenberg, B.; Zimmermann, N. (2003). "Iridium-Catalyzed Enantioselective Hydrogenation of Olefins". Advanced Synthesis & Catalysis. 345 (12): 33. doi:10.1002/adsc.200390027.
- ^ Cui, X.; Burgess, K. (2005). "Catalytic Homogeneous Asymmetric Hydrogenations of Largely Unfunctionalized Alkenes". Chemical Reviews. 105 (9): 3272–3296. doi:10.1021/cr0500131. PMID 16159153.
- ^ a b Cite error: The named reference
IrKetoneswas invoked but never defined (see the help page). - ^ Blaser, H. U.; Pugin, B. T.; Spindler, F.; Togni, A. (2002). "Enantioselective imine hydrogenation with Ir diphosphine catalysts: Fighting deactivation". Comptes Rendus Chimie. 5 (5): 379. doi:10.1016/S1631-0748(02)01391-7.
- ^ Enthaler, S.; Junge, K.; Beller, M. (2008). "Sustainable Metal Catalysis with Iron: From Rust to a Rising Star?". Angewandte Chemie International Edition. 47 (18): 3317–21. doi:10.1002/anie.200800012. PMID 18412184.
- ^ Whitesell, J. K. (1989). "C2 symmetry and asymmetric induction". Chemical Reviews. 89 (7): 1581–1590. doi:10.1021/cr00097a012.
- ^ Knowles, W. S.; Sabacky, M. J.; Vineyard, B. D. (1972). "Catalytic asymmetric hydrogenation". Journal of the Chemical Society, Chemical Communications (1): 10. doi:10.1039/C39720000010. PMID 4270504.
- ^ a b Jerphagnon, T.; Renaud, J. L.; Bruneau, C. (2004). "Chiral monodentate phosphorus ligands for rhodium-catalyzed asymmetric hydrogenation". Tetrahedron: Asymmetry. 15 (14): 2101. doi:10.1016/j.tetasy.2004.04.037.
- ^ Vineyard, B. D.; Knowles, W. S.; Sabacky, M. J.; Bachman, G. L.; Weinkauff, D. J. (1977). "Asymmetric hydrogenation. Rhodium chiral bisphosphine catalyst". Journal of the American Chemical Society. 99 (18): 5946. doi:10.1021/ja00460a018.
- ^ Knowles, W. S.; Sabacky, M. J.; Vineyard, B. D.; Weinkauff, D. J. (1975). "Asymmetric hydrogenation with a complex of rhodium and a chiral bisphosphine". Journal of the American Chemical Society. 97 (9): 2567. doi:10.1021/ja00842a058.
- ^ Müller, D.; Umbricht, G.; Weber, B.; Pfaltz, A. (1991). "C2-Symmetric 4,4',5,5'-Tetrahydrobi(oxazoles) and 4,4',5,5'-Tetrahydro-2,2'-methylenebis[oxazoles] as Chiral Ligands for Enantioselective Catalysis Preliminary Communication". Helvetica Chimica Acta. 74: 232–240. doi:10.1002/hlca.19910740123.
- ^ Helmchen, G. N.; Pfaltz, A. (2000). "PhosphinooxazolinesA New Class of Versatile, Modular P,N-Ligands for Asymmetric Catalysis". Accounts of Chemical Research. 33 (6): 336–345. doi:10.1021/ar9900865. PMID 10891051.
- ^ Cite error: The named reference
IrOlefinswas invoked but never defined (see the help page). - ^ Noyori, R.; Ohkuma, T. (2001). "Asymmetric Catalysis by Architectural and Functional Molecular Engineering: Practical Chemo- and Stereoselective Hydrogenation of Ketones". Angewandte Chemie International Edition. 40 (1): 40–73. doi:10.1002/1521-3773(20010105)40:1<40::AID-ANIE40>3.0.CO;2-5. PMID 11169691.
- ^ Hems, W. P.; Groarke, M.; Zanotti-Gerosa, A.; Grasa, G. A. (2007). "[(Bisphosphine) Ru(II) Diamine] Complexes in Asymmetric Hydrogenation: Expanding the Scope of the Diamine Ligand". Accounts of Chemical Research. 40 (12): 1340–1347. doi:10.1021/ar7000233. PMID 17576143.
- ^ Cite error: The named reference
IrRigidwas invoked but never defined (see the help page). - ^ a b Zhou, Y. G. (2007). "Asymmetric Hydrogenation of Heteroaromatic Compounds". Accounts of Chemical Research. 40 (12): 1357–1366. CiteSeerX 10.1.1.653.5495. doi:10.1021/ar700094b. PMID 17896823.
- ^ Lam, K. H.; Xu, L.; Feng, L.; Fan, Q. H.; Lam, F. L.; Lo, W. H.; Chan, A. S. C. (2005). "Highly Enantioselective Iridium-Catalyzed Hydrogenation of Quinoline Derivatives Using Chiral Phosphinite H8-BINAPO". Advanced Synthesis & Catalysis. 347 (14): 1755. doi:10.1002/adsc.200505130.
- ^ Ye, Z. S.; Chen, M. W.; Chen, Q. A.; Shi, L.; Duan, Y.; Zhou, Y. G. (2012). "Iridium-Catalyzed Asymmetric Hydrogenation of Pyridinium Salts". Angewandte Chemie International Edition. 51 (40): 10181–4. doi:10.1002/anie.201205187. PMID 22969060.
- ^ Cite error: The named reference
OrganocatalyticIndoleswas invoked but never defined (see the help page). - ^ a b Kuwano, R.; Kashiwabara, M.; Ohsumi, M.; Kusano, H. (2008). "Catalytic Asymmetric Hydrogenation of 2,3,5-Trisubstituted Pyrroles". Journal of the American Chemical Society. 130 (3): 808–809. doi:10.1021/ja7102422. PMID 18154340.
- ^ a b Wang, D. S.; Ye, Z. S.; Chen, Q. A.; Zhou, Y. G.; Yu, C. B.; Fan, H. J.; Duan, Y. (2011). "Highly Enantioselective Partial Hydrogenation of Simple Pyrroles: A Facile Access to Chiral 1-Pyrrolines". Journal of the American Chemical Society. 133 (23): 8866–8869. doi:10.1021/ja203190t. PMID 21591641.
- ^ Wang, D. S.; Chen, Q. A.; Lu, S. M.; Zhou, Y. G. (2012). "Asymmetric Hydrogenation of Heteroarenes and Arenes". Chemical Reviews. 112 (4): 2557–2590. doi:10.1021/cr200328h. PMID 22098109.
- ^ Urban, S.; Beiring, B.; Ortega, N.; Paul, D.; Glorius, F. (2012). "Asymmetric Hydrogenation of Thiophenes and Benzothiophenes". Journal of the American Chemical Society. 134 (37): 15241–15244. doi:10.1021/ja306622y. PMID 22934527.
- ^ Heitbaum, M.; Glorius, F.; Escher, I. (2006). "Asymmetric Heterogeneous Catalysis". Angewandte Chemie International Edition. 45 (29): 4732–62. doi:10.1002/anie.200504212. PMID 16802397.
- ^ Blaser, H. U.; Spindler, F.; Studer, M. (2001). "Enantioselective catalysis in fine chemicals production". Applied Catalysis A: General. 221 (1–2): 119–143. doi:10.1016/S0926-860X(01)00801-8. PMID 12613584.
- ^ Dub, Pavel A.; Gordon, John C. (2018). "The role of the metal-bound N–H functionality in Noyori-type molecular catalysts". Nature Reviews Chemistry. 2 (12): 396–408. doi:10.1038/s41570-018-0049-z. S2CID 106394152.
- ^ Blaser, Hans-Ulrich; Federsel, Hans-Jürgen, eds. (2010). Asymmetric Catalysis on Industrial Scale. Weinheim: Wiley-VCH. pp. 13–16. doi:10.1002/9783527630639. ISBN 978-3-527-63063-9.
- ^ Jacobsen, E.N.; Pfaltz, Andreas; Yamamato, H., eds. (1999). Comprehensive Asymmetric Catalysis. Berlin; New York: Springer. pp. 1443–1445. ISBN 978-3-540-64336-4.
Category:Organic reactions Category:Chemical processes Category:Green chemistry Category:Hydrogenation Hydrogenation