
Vacuum distillation

Vacuum distillation or distillation under reduced pressure is a type of distillation performed under reduced pressure, which allows the purification of compounds not readily distilled at ambient pressures or simply to save time or energy. This technique separates compounds based on differences in their boiling points. This technique is used when the boiling point of the desired compound is difficult to achieve or will cause the compound to decompose.[1] Reduced pressures decrease the boiling point of compounds. The reduction in boiling point can be calculated using a temperature-pressure nomograph using the Clausius–Clapeyron relation.[2]
Laboratory-scale applications
[edit]Compounds with a boiling point lower than 150 °C typically are distilled at ambient pressure. For samples with high boiling points, short-path distillation apparatus is commonly employed.[3][4] This technique is amply illustrated in Organic Synthesis.[5] [6]
Rotary evaporation
[edit]Rotary evaporation[7] is a common technique used in laboratories to concentrate or isolate a compound from solution. Many solvents are volatile and can easily be evaporated using rotary evaporation. Even less volatile solvents can be removed by rotary evaporation under high vacuum and with heating. It is also used by environmental regulatory agencies for determining the amount of solvents in paints, coatings and inks.[8]
Safety considerations
[edit]Safety is an important consideration when glassware is under vacuum pressure. Scratches and cracks can result in implosions when the vacuum is applied. Wrapping as much of the glassware with tape as is practical helps to prevent dangerous scattering of glass shards in the event of an implosion.[citation needed]
Industrial-scale applications
[edit]

Industrial-scale vacuum distillation[10] has several advantages. Close boiling mixtures may require many equilibrium stages to separate the key components. One tool to reduce the number of stages needed is to utilize vacuum distillation.[11] Vacuum distillation columns (as depicted in Figures 2 and 3) typically used in oil refineries have diameters ranging up to about 14 meters (46 feet), heights ranging up to about 50 meters (164 feet), and feed rates ranging up to about 25,400 cubic meters per day (160,000 barrels per day).[citation needed]
Vacuum distillation can improve a separation by:[citation needed]
- Prevention of product degradation or polymer formation because of reduced pressure leading to lower tower bottoms temperatures,
- Reduction of product degradation or polymer formation because of reduced mean residence time especially in columns using packing rather than trays.
- Increasing capacity, yield, and purity.
Another advantage of vacuum distillation is the reduced capital cost, at the expense of slightly more operating cost. Utilizing vacuum distillation can reduce the height and diameter, and thus the capital cost of a distillation column.[citation needed]
Vacuum distillation in petroleum refining
[edit]Petroleum crude oil is a complex mixture of hundreds of different hydrocarbon compounds generally having from 3 to 60 carbon atoms per molecule, although there may be small amounts of hydrocarbons outside that range.[12][13][14] The refining of crude oil begins with distilling the incoming crude oil in a so-called atmospheric distillation column operating at pressures slightly above atmospheric pressure.[10][12][13]
Vacuum distillation can also be referred to as "low-temperature distillation".[citation needed]
In distilling the crude oil, it is important not to subject the crude oil to temperatures above 370 to 380 °C because high molecular weight components in the crude oil will undergo thermal cracking and form petroleum coke at temperatures above that. Formation of coke would result in plugging the tubes in the furnace that heats the feed stream to the crude oil distillation column. Plugging would also occur in the piping from the furnace to the distillation column as well as in the column itself.[citation needed]
The constraint imposed by limiting the column inlet crude oil to a temperature of less than 370 to 380 °C yields a residual oil from the bottom of the atmospheric distillation column consisting entirely of hydrocarbons that boil above 370 to 380 °C.
To further distill the residual oil from the atmospheric distillation column, the distillation must be performed at absolute pressures as low as 10 to 40 mmHg / Torr (About 5% atmospheric pressure) so as to limit the operating temperature to less than 370 to 380 °C.
Figure 2 is a simplified process diagram of a petroleum refinery vacuum distillation column that depicts the internals of the column and Figure 3 is a photograph of a large vacuum distillation column in a petroleum refinery.
The 10 to 40 mmHg absolute pressure in a vacuum distillation column increases the volume of vapor formed per volume of liquid distilled. The result is that such columns have very large diameters.[15]
Distillation columns such those in Images 1 and 2, may have diameters of 15 meters or more, heights ranging up to about 50 meters, and feed rates ranging up to about 25,400 cubic meters per day (160,000 barrels per day).
The vacuum distillation column internals must provide good vapor–liquid contacting while, at the same time, maintaining a very low-pressure increase from the top of the column top to the bottom. Therefore, the vacuum column uses distillation trays only where products are withdrawn from the side of the column (referred to as side draws). Most of the column uses packing material for the vapor–liquid contacting because such packing has a lower pressure drop than distillation trays. This packing material can be either structured sheet metal or randomly dumped packing such as Raschig rings.
The absolute pressure of 10 to 40 mmHg in the vacuum column is most often achieved by using multiple stages of steam jet ejectors.[16]
Many industries, other than the petroleum refining industry, use vacuum distillation on a much smaller scale. Copenhagen-based Empirical Spirits,[17] a distillery founded by former Noma chefs,[18] uses the process to create uniquely flavoured spirits. Their flagship spirit, Helena, is created using Koji, alongside Pilsner Malt and Belgian Saison Yeast.[19]
Large-scale water purification
[edit]Vacuum distillation is often used in large industrial plants as an efficient way to remove salt from ocean water, in order to produce fresh water. This is known as desalination. The ocean water is placed under a vacuum to lower its boiling point and has a heat source applied, allowing the fresh water to boil off and be condensed. The condensing of the water vapor prevents the water vapor from filling the vacuum chamber, and allows the effect to run continuously without a loss of vacuum pressure. The heat from condensation of the water vapor is removed by a heat sink, which uses the incoming ocean water as the coolant and thus preheats the feed of ocean water. Some forms of distillation do not use condensers, but instead compress the vapor mechanically with a pump. This acts as a heat pump, concentrating the heat from the vapor and allowing for the heat to be returned and reused by the incoming untreated water source. There are several forms of vacuum distillation of water, with the most common being multiple-effect distillation, vapor-compression desalination, and multi-stage flash distillation.[20]
Molecular distillation
[edit]Molecular distillation is vacuum distillation below the pressure of 0.01 torr[21] (1.3 Pa). 0.01 torr is one order of magnitude above high vacuum, where fluids are in the free molecular flow regime, i.e. the mean free path of molecules is comparable to the size of the equipment.[1] The gaseous phase no longer exerts significant pressure on the substance to be evaporated, and consequently, the rate of evaporation no longer depends on pressure. That is, because the continuum assumptions of fluid dynamics no longer apply, mass transport is governed by molecular dynamics rather than fluid dynamics. Thus, a short path between the hot surface and the cold surface is necessary, typically by suspending a hot plate covered with a film of feed next to a cold plate with a line of sight in between.[citation needed]
Molecular distillation is used industrially for purification of oils.[20]
Gallery
[edit]-
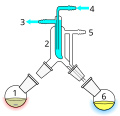 A simple short path vacuum distillation apparatus
A simple short path vacuum distillation apparatus -
![Kugelrohr – a short path[1] vacuum distillation apparatus](//upload.wikimedia.org/wikipedia/commons/thumb/8/82/Kugelrohr_-_numbered.jpg/120px-Kugelrohr_-_numbered.jpg)
-
 Perkin triangle – for air-sensitive vacuum distillation
Perkin triangle – for air-sensitive vacuum distillation -
 Vacuum distillation apparatus
Vacuum distillation apparatus

![Kugelrohr – a short path[1] vacuum distillation apparatus](/wiki/File:Kugelrohr_-_numbered.jpg)


See also
[edit]References
[edit]- ^ a b c Hickman, K. C. D. (1945), "Adventures in vacuum chemistry", American Scientist, 33 (4), Scientific american: xxx–231, JSTOR 27826079, retrieved 2021-09-01
- ^ "Pressure-Temperature Nomograph Interactive Tool". Sigma-Aldrich. Retrieved 2018-03-23.
- ^ Introduction to Organic Laboratory Techniques: A Small Scale Approach By Donald L. Pavia, Gary M. Lampman, George S. Kriz, Randall G. Engel. Chapter 16.
- ^ Leonard, J.; Lygo, B.; Procter, Garry (8 January 2013). Advanced practical organic chemistry (3rd ed.). Boca Raton. ISBN 9781439860977. OCLC 883131986.
{{cite book}}: CS1 maint: location missing publisher (link) - ^ Fastuca, Nicholas J.; Wong, Alice R.; Mak, Victor W.; Reisman, Sarah E. (2020). "Asymmetric Michael Addition of Dimethyl Malonate to 2 Cyclopenten-1-one Catalyzed by a Heterobimetallic Complex". Organic Syntheses. 97: 327–338. doi:10.15227/orgsyn.097.0327. PMC 9128456. PMID 35614904.
- ^ Bartko, Samuel G.; Deng, James; Danheiser, Rick L. (2016). "Synthesis of 1-Iodopropyne". Organic Syntheses. 93: 245–262. doi:10.15227/orgsyn.093.0245.
- ^ Operation of a Rotary Evaporator (Rotovap) (from the website of the University of British Columbia)
- ^ [1] SCAQMD Test method 302-91
- ^ Energy Institute website page
- ^ a b Kister, Henry Z. (1992). Distillation Design (1st ed.). McGraw-Hill. ISBN 0-07-034909-6.
- ^ Karl Kolmetz, Andrew W. Sloley et al. (2004), Designing Distillation Columns for Vacuum Service, 11th India Oil and Gas Symposium and International Exhibition, September 2004, Mumbai, India (also published in Hydrocarbon Processing, May 2005)
- ^ a b Gary, J.H. & Handwerk, G.E. (1984). Petroleum Refining Technology and Economics (2nd ed.). Marcel Dekker, Inc. ISBN 0-8247-7150-8.
- ^ a b Leffler, W.L. (1985). Petroleum refining for the nontechnical person (2nd ed.). PennWell Books. ISBN 0-87814-280-0.
- ^ James G, Speight (2006). The Chemistry and Technology of Petroleum (4th ed.). CRC Press. 0-8493-9067-2.
- ^ Karl Kolmetz, Andrew W. Sloley et al (2004), Designing Distillation Columns for Vacuum Service, 11th India Oil and Gas Symposium and International Exhibition, September 2004, Mumbai, India (also published in Hydrocarbon Processing, May 2005)
- ^ Photo gallery Archived 2009-02-07 at the Wayback Machine (from website of Graham Manufacturing Company)
- ^ "Empirical Spirits". empiricalspirits.co. Retrieved 2018-10-15.
- ^ Kahn, Howie (2018-04-25). "A Fresh Take on Liquor From Two Noma Alums". Wall Street Journal. ISSN 0099-9660. Retrieved 2018-10-15.
- ^ "Empirical Spirits". Empirical Spirits. Retrieved 2018-10-15.
- ^ a b Desalination and Water Treatment, Murat Eyvaz, Ebubekir Yüksel 1988/2018. Chapter 5.
- ^ Vogel's 5th ed.
- This article incorporates material from the Citizendium article "Vacuum distillation", which is licensed under the Creative Commons Attribution-ShareAlike 3.0 Unported License but not under the GFDL.
External links
[edit]- D1160 Vacuum Distillation
- How vacuum distillation works
- Pressure-temperature nomograph
- Short path distillation, includes a table comparing methods
| Principles |  | |
|---|---|---|
| Industrial processes | ||
| Laboratory methods | ||
| Techniques | ||