
Simmons–Smith reaction
| Simmons-Smith reaction | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Named after | Howard Ensign Simmons, Jr. Ronald D. Smith | ||||||||
| Reaction type | Ring forming reaction | ||||||||
| Reaction | |||||||||
| |||||||||
| Conditions | |||||||||
| Identifiers | |||||||||
| Organic Chemistry Portal | simmons-smith-reaction | ||||||||
| RSC ontology ID | RXNO:0000258 | ||||||||
The Simmons–Smith reaction is an organic cheletropic reaction involving an organozinc carbenoid that reacts with an alkene (or alkyne) to form a cyclopropane.[1][2][3] It is named after Howard Ensign Simmons, Jr. and Ronald D. Smith. It uses a methylene free radical intermediate that is delivered to both carbons of the alkene simultaneously, therefore the configuration of the double bond is preserved in the product and the reaction is stereospecific.[4]

Mechanism[edit]
 Simmons-Smith reaction in progress |

Examples[edit]
Thus, cyclohexene, diiodomethane, and a zinc-copper couple (as iodomethylzinc iodide, ICH2ZnI) yield norcarane (bicyclo[4.1.0]heptane).[5][6]

The Simmons–Smith reaction is generally preferred over other methods of cyclopropanation,[7] however it can be expensive due to the high cost of diiodomethane. Modifications involving cheaper alternatives have been developed, such as dibromomethane[8] or diazomethane and zinc iodide.[9] The reactivity of the system can also be increased by using the Furukawa modification, exchanging the zinc‑copper couple for diethylzinc.[10]
The Simmons–Smith reaction is generally subject to steric effects, and thus cyclopropanation usually takes place on the less hindered face.[11][12] However, when a hydroxy substituent is present in the substrate in proximity to the double bond, the zinc coordinates with the hydroxy substituent, directing cyclopropanation cis to the hydroxyl group (which may not correspond to cyclopropanation of the sterically most accessible face of the double bond):[13] An interactive 3D model of this reaction can be seen at ChemTube3D.

Asymmetric Simmons–Smith reaction[edit]
Although asymmetric cyclopropanation methods based on diazo compounds (the Metal-catalyzed cyclopropanations) exist since 1966, the asymmetric Simmons–Smith reaction was introduced in 1992 [14] with a reaction of cinnamyl alcohol with diethylzinc, diiodomethane and a chiral disulfonamide in dichloromethane:

The hydroxyl group is a prerequisite serving as an anchor for zinc. An interactive 3D model of a similar reaction[15] can be seen here (java required). In another version of this reaction the ligand is based on salen and Lewis acid DIBAL is added:[16]

Scope and limitations[edit]
Achiral alkenes[edit]
The Simmons–Smith reaction can be used to cyclopropanate simple alkenes without complications. Unfunctionalized achiral alkenes are best cyclopropanated with the Furukawa modification (see below), using Et2Zn and CH2I2 in 1,2-dichloroethane.[17] Cyclopropanation of alkenes activated by electron donating groups proceed rapidly and easily. For example, enol ethers like trimethylsilyloxy-substituted olefins are often used because of the high yields obtained.[18]

Despite the electron-withdrawing nature of halides, many vinyl halides are also easily cyclopropanated, yielding fluoro-, bromo-, and iodo-substituted cyclopropanes.[19][20]
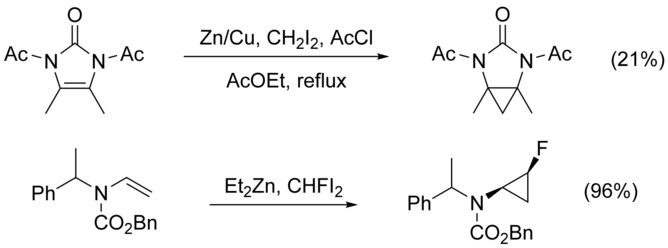
The cyclopropanation of N-substituted alkenes is made complicated by N-alkylation as a competing pathway. This can be circumvented by adding a protecting group to nitrogen, however the addition of electron-withdrawing groups decreases the nucleophilicity of the alkene, lowering yield. The use of highly electrophilic reagents such as CHFI2, in place of CH2I2, has been shown to increase yield in these cases.[21]
Polyenes[edit]
Without the presence of a directing group on the olefin, very little chemoselectivity is observed.[22] However, an alkene which is significantly more nucleophilic than any others will be highly favored. For example, cyclopropanation occurs highly selectively at enol ethers.[23]

Functional group compatibility[edit]
An important aspect of the Simmons–Smith reaction that contributes to its wide usage is its ability to be used in the presence of many functional groups. Among others, the haloalkylzinc-mediated reaction is compatible with alkynes, alcohols, ethers, aldehydes, ketones, carboxylic acids and derivatives, carbonates, sulfones, sulfonates, silanes, and stannanes. However, some side reactions are commonly observed.
Most side reactions occur due to the Lewis-acidity of the byproduct, ZnI2. In reactions that produce acid-sensitive products, excess Et2Zn can be added to scavenge the ZnI2 that is formed, forming the less acidic EtZnI. The reaction can also be quenched with pyridine, which will scavenge ZnI2 and excess reagents.[24]
Methylation of heteroatoms is also observed in the Simmons–Smith reaction due to the electrophilicity of the zinc carbenoids. For example, the use of excess reagent for long reaction times almost always leads to the methylation of alcohols.[25] Furthermore, Et2Zn and CH2I2 react with allylic thioethers to generate sulfur ylides, which can subsequently undergo a 2,3-sigmatropic rearrangement, and will not cyclopropanate an alkene in the same molecule unless excess Simmons–Smith reagent is used.[26]

Modifications[edit]
While the Simmons–Smith reaction is often discussed in its basic form, a number of modifications to both the zinc catalyst and the added carbon have been proposed.
Furukawa modification[edit]
The Furukawa modification involves the replacement of the zinc-copper couple with dialkyl zinc, the most active of which was found to be Et2Zn. The modification was proposed in 1968 as a way to turn cationically polymerizable olefins such as vinyl ethers into their respective cyclopropanes.[27] It has also been found to be especially useful for the cyclopropanation of carbohydrates, being far more reproducible than other methods.[28] Like the unmodified reaction, the Furukawa-modified reaction is stereospecific, and is often much faster than the unmodified reaction. However, the Et2Zn reagent is pyrophoric, and as such must be handled with care.[29]

Charette modification[edit]
The Charette modification replaces the CH2I2 normally found in the Simmons–Smith reaction with aryldiazo compounds, such as phenyldiazomethane, in Pathway A.[30] Upon treatment with stoichiometric amounts of zinc halide, an organozinc compound similar to the carbenoid discussed above is produced. This can react with almost all alkenes and alkynes, including styrenes and alcohols. This is especially useful, as the unmodified Simmons-Smith is known to deprotonate alcohols. Unfortunately, as in Pathway B shown the intermediate can also react with the starting diazo compound, giving cis- or trans- 1,2-diphenylethene. Additionally, the intermediate can react with alcohols to produce iodophenylmethane, which can further undergo an SN2 reaction to produce ROCHPh, as in Pathway C.

Non-zinc reagents[edit]
Although not commonly used, Simmons-Smith reagents that display similar reactive properties to those of zinc have been prepared from aluminum and samarium compounds in the presence of CH2IX.[31] With the use of these reagents, allylic alcohols and isolated olefins can be selectively cyclopropanated in the presence of each other. Iodo- or chloro- methylsamarium iodide in THF is an excellent reagent to selectively cyclopropanate the allylic alcohol, presumably directed by chelation to the hydroxyl group.[32] In contrast, use of dialkyl(iodomethyl)aluminum reagents in CH2Cl2 will selectively cyclopropanate the isolated olefin.[33] The specificity of these reagents allow cyclopropanes to be placed in poly-unsaturated systems that zinc-based reagents will cyclopropanate fully and unselectively. For example, i-Bu3Al will cyclopropanate geraniol at the 6 position, while Sm/Hg, will cyclopropanate at the 2 position, as shown below.

However, both reactions require near stoichiometric amounts of the starting metal compound, and Sm/Hg must be activated with the highly toxic HgCl2.
Uses in synthesis[edit]
Most modern applications of the Simmons–Smith reaction use the Furukawa modification. Especially relevant and reliable applications are listed below.
Insertion to form γ-keto esters[edit]
A Furukawa-modified Simmons-Smith generated cyclopropane intermediate is formed in the synthesis of γ-keto esters from β-keto esters. The Simmons-Smith reagent binds first to the carbonyl group and subsequently to the α-carbon of the pseudo-enol that the first reaction forms. This second reagent forms the cyclopropyl intermediate which rapidly fragments into the product.[34][35]

Formation of amido-spiro [2.2] pentanes from allenamides[edit]
A Furukawa-modified Simmons–Smith reaction cyclopropanates both double bonds in an allenamide to form amido-spiro [2.2] cyclopentanes, featuring two cyclopropyl rings which share one carbon. The product of monocyclopropanation is also formed.[36][37]
_pentane_2.png)
Natural product synthesis[edit]
Cyclopropanation reactions in natural products synthesis have been reviewed.[38] The β-lactamase inhibitor Cilastatin provides an instructive example of Simmons-Smith reactivity in natural products synthesis. An allyl substituent on the starting material is Simmons-Smith cyclopropanated, and the carboxylic acid is subsequently deprotected via ozonolysis to form the precursor.


Drugs synthesis[edit]
The Simmons–Smith reaction is used in the syntheses of GSK1360707F and ropanicant.[39]
References[edit]
- ^ Howard Ensign Simmons Jr.; Smith, R.D. (1958). "A New Synthesis of Cyclopropanes from Olefins". J. Am. Chem. Soc. 80 (19): 5323–5324. doi:10.1021/ja01552a080.
- ^ Simmons, H.E.; Smith, R.D. (1959). "A New Synthesis of Cyclopropanes". J. Am. Chem. Soc. 81 (16): 4256–4264. doi:10.1021/ja01525a036.
- ^ Denis, J.M.; Girard, J.M.; Conia, J.M (1972). "Improved Simmons–Smith Reactions". Synthesis. 1972 (10): 549–551. doi:10.1055/s-1972-21919.
- ^ Charette, A. B.; Beauchemin, A. (2001). "Simmons-Smith Cyclopropanation Reaction". Org. React. 58: 1. doi:10.1002/0471264180.or058.01. ISBN 978-0-471-26418-7.
- ^ Smith, R. D.; Simmons, H. E. "Norcarane". Organic Syntheses
{{cite journal}}: CS1 maint: multiple names: authors list (link); Collected Volumes, vol. 5, p. 855. - ^ Ito, Y.; Fujii, S.; Nakatuska, M.; Kawamoto, F.; Saegusa, T. (1988). "One-Carbon Ring Expansion Of Cycloalkanones To Conjugated Cycloalkenones: 2-Cyclohepten-1-one". Organic Syntheses
{{cite journal}}: CS1 maint: multiple names: authors list (link); Collected Volumes, vol. 6, p. 327. - ^ Clayden, Jonathan; Greeves, Nick; Warren, Stuart; Wothers, Peter (2001). Organic Chemistry (1st ed.). Oxford University Press. ISBN 978-0-19-850346-0.Page 1067
- ^ Fabisch, Bodo; Mitchell, Terence N. (1984). "An inexpensive modification of the Simmons-Smith reaction: The formation of bromomethylzinc bromide as studied by NMR spectroscopy". Journal of Organometallic Chemistry. 269 (3): 219–221. doi:10.1016/0022-328X(84)80305-8.
- ^ Wittig, Georg; Wingler, Frank (1 August 1964). "Über methylenierte Metallhalogenide, IV. Cyclopropan-Bildung aus Olefinen mit Bis-halogenmethyl-zink". Chemische Berichte. 97 (8): 2146–2164. doi:10.1002/cber.19640970808.
- ^ Furukawa, J.; Kawabata, N.; Nishimura, J. (1968). "Synthesis of cyclopropanes by the reaction of olefins with dialkylzinc and methylene iodide". Tetrahedron. 24 (1): 53–58. doi:10.1016/0040-4020(68)89007-6.
- ^ Simmons, Howard E.; Cairns, Theodore L.; Vladuchick, Susan A.; Hoiness, Connie M. (2011-03-15), "Cyclopropanes from Unsaturated Compounds, Methylene Iodide, and Zinc-Copper Couple", Organic Reactions, Hoboken, NJ, USA: John Wiley & Sons, Inc., pp. 1–131, doi:10.1002/0471264180.or020.01, ISBN 978-0-471-26418-7, retrieved 2022-02-28
- ^ Girard, C.; Conia, J. M. (1978). Journal of Chemical Research, Synopses (Review): 182–.
{{cite journal}}: Missing or empty|title=(help) - ^ Paul A. Grieco; Tomei Oguri; Chia-Lin J. Wang & Eric Williams (1977). "Stereochemistry and total synthesis of (±)-ivangulin". J. Org. Chem. 42 (25): 4113–4118. doi:10.1021/jo00445a027.
- ^ Hideyo Takahashi, Masato Yoshioka, Masaji Ohno and Susumu Kobayashi (1992). "A catalytic enantioselective reaction using a C2-symmetric disulfonamide as a chiral ligand: cyclopropanation of allylic alcohols by the Et2Zn-CH2I2-disulfonamide system". Tetrahedron Letters. 33 (18): 2575–2578. doi:10.1016/S0040-4039(00)92246-9.
{{cite journal}}: CS1 maint: multiple names: authors list (link) - ^ Wang, Tao; Liang, Yong; Yu, Zhi-Xiang (2011). "Density Functional Theory Study of the Mechanism and Origins of Stereoselectivity in the Asymmetric Simmons–Smith Cyclopropanation with Charette Chiral Dioxaborolane Ligand". Journal of the American Chemical Society. 133 (24): 9343–9353. doi:10.1021/ja111330z. PMID 21627114.
- ^ Hiroaki Shitama & Tsutomu Katsuki (2008). "Asymmetric Simmons–Smith Reaction of Allylic Alcohols with Al Lewis Acid/N Lewis Base Bifunctional Al(Salalen) Catalyst". Angew. Chem. Int. Ed. 47 (13): 2450–2453. doi:10.1002/anie.200705641. PMID 18288666.
- ^ Denmark, S. E.; Edwards, J. P. (1991). "A Comparison of (Chloromethyl)- and (Iodomethyl)zinc Cyclopropanation Reagents". J. Org. Chem. 56 (25): 6974–6981. doi:10.1021/jo00025a007.
- ^ Rubottom, G. M.; Lopez, M. I. (1973). "Reaction of Trimethysilyl Enol Ethers with Simmons-Smith Reagent. Facile Synthesis of Trimethylsilyl Cyclopropyl Ethers and Cyclopropanols". J. Org. Chem. 38 (11): 2097–2099. doi:10.1021/jo00951a032.
- ^ Morikawa, T.; Sasaki, H.; Mori, K.; Shiro, M.; Taguchi, T.; Morikawa, T.; Sasaki, H.; Mori, K.; Shiro, M.; Taguchi, T. Simmons-Smith Reactions of Fluoroallyl Alcohol Derivatives. Chem. Pharm. Bull. (Tokyo) 1992, 40 (12), 3189.
- ^ Piers, E.; Coish, P. D. Preparation and Cyclopropanation of 2- and 3-Iodoalk-2-En-1-Ols: Synthesis of Functionalized, Stereodefined Iodocyclopropanes. Synthesis 1995, 1995 (1), 47–55.
- ^ Gagnon, J. L.; Jr, W. W. Z. Synthesis of Cis-1,5-Dimethyl-2,4-Dinitro-2,4-diazabicyclo[3.1.0]hexan-3-One and Cis-1,5-Dimethyl-2,4-Dinitro-2,4-diazabicyclo[3.2.0]heptan-3-One. Synth. Commun. 1996, 26 (4), 837–845.
- ^ Friedrich, E. C.; Niyati-Shirkhodaee, F. (1991). "Regioselectivity and Solvent Effects in Cyclopropanation of Alkadienes". J. Org. Chem. 56 (6): 2202–2205. doi:10.1021/jo00006a044.
- ^ Lee, J.; Kim, H.; Cha, J. K. (1995). "Diastereoselective Synthesis of Cis-1,2-Dialkenylcyclopropanols and Subsequent Oxy-Cope Rearrangement". J. Am. Chem. Soc. 117 (39): 9919–9920. doi:10.1021/ja00144a022.
- ^ Denis, J. M.; Girard, C.; Conia, J. M. Improved Simmons-Smith Reactions. Synthesis 1972, 1972 (10), 549–551.
- ^ Takakis, I. M.; Rhodes, Y. E. (1978). "Cyclopropanation of Some Simple Olefinic Compounds. By-Product Formation in Excess Simmons-Smith Reagent". J. Org. Chem. 43 (18): 3496–3500. doi:10.1021/jo00412a017.
- ^ Cohen, T.; Kosarych, Z. (1982). "Complete regio- and stereospecificity in the Lewis acid catalyzed Diels-Alder reactions of (Z)-2-methoxy-1-(phenylthio)-1,3-butadienes. Conversion of the CS configuration of an adduct to the CC configuration at the allylic position by a [2,3] sigmatropic rearrangement". J. Org. Chem. 47 (20): 4005–4008. doi:10.1021/jo00141a047.
- ^ Furukawa, J; Kawabata, N; Nishimura, J (1968). "Synthesis of cyclopropanes by the reaction of olefins with dialkylzinc and methylene iodide". Tetrahedron. 24 (1): 53–58. doi:10.1016/0040-4020(68)89007-6.
- ^ Halton, B (2000). Advances in Strained and Interesting Organic Molecules, Volume 8. Stamford, Ct: Press Inc. p. 115. ISBN 978-0-7623-0631-2.
- ^ "Diethyl Zinc MSDS" (PDF). Retrieved 10 May 2017.
- ^ Lévesque, Éric; Goudreau, Sébastien R.; B. Charette, André B. (2014). "Improved Zinc-Catalyzed Simmons–Smith Reaction: Access to Various 1,2,3-Trisubstituted Cyclopropanes". Organic Letters. 16 (5): 1490–1493. doi:10.1021/ol500267w. PMID 24555697.
- ^ Roger, Adams (2001). Organic Reactions Vol 58. New York: Wiley, J. pp. 9–10. ISBN 978-0-471-10590-9.
- ^ Molander, G. A.; Harring, L. S. (1989). "Samarium-Promoted Cyclopropanation of Allylic Alcohols". J. Org. Chem. 54 (15): 3525–3532. doi:10.1021/jo00276a008.
- ^ Maruoka, K.; Fukutani, Y.; Yamamoto, H. (1985). "Trialkylaluminum-Alkylidene Iodide. A Powerful Cyclopropanation Agent with Unique Selectivity". J. Org. Chem. 50 (22): 4412–4414. doi:10.1021/jo00222a051.
- ^ Bhogadhi, Yashoda; Zercher, Charles (2014). "Discussion Addendum for: Formation of γ-Keto Esters from β-Keto Esters: Methyl 5,5-dimethyl-4-oxohexanoate". Organic Syntheses. 91: 248–259. doi:10.15227/orgsyn.091.0248.
- ^ Ronsheim, Matthew; Hilgenkamp, Ramona; Zercher, Charles (2002). "Formation of γ-Keto Esters from β-Keto Esters: Methyl 5,5-dimethyl-4-oxohexanoate". Organic Syntheses. 79: 146. doi:10.1002/0471264180.os079.18.
- ^ Teo, Yong-Chua; Hsung, Richard (2014). "Discussion Addendum for: Practical Synthesis of Novel Chiral Allenamides: (R)-4- Phenyl-3-(1,2-propadienyl)oxazolidin-2-one". Organic Syntheses. 91: 12–26. doi:10.15227/orgsyn.091.0012.
- ^ Xiong, H; Tracey, M; Grebe, T; Mulder, J; Hsung, R (2005). "PRACTICAL SYNTHESIS OF NOVEL CHIRAL ALLENAMIDES: (R)-4-PHENYL-3-(1,2-PROPADIENYL)OXAZOLIDIN-2-ONE (2-Oxazolidinone, 4-phenyl-3-(1,2-propadienyl)–, (4R)–)". Organic Syntheses. 81: 147–156. doi:10.15227/orgsyn.081.0147.
- ^ Donaldson, William (October 8, 2001). "Synthesis of cyclopropane containing natural products". Tetrahedron. 57 (41): 8589. doi:10.1016/s0040-4020(01)00777-3.
- ^ "Synthesis of SUVN-911". Synfacts. 16 (6): 0626. 2020. doi:10.1055/s-0040-1707534. ISSN 1861-1958. S2CID 219767881.
External links[edit]