
Wacker process

The Wacker process or the Hoechst-Wacker process (named after the chemical companies of the same name) refers to the oxidation of ethylene to acetaldehyde in the presence of palladium(II) chloride and copper(II) chloride as the catalyst.[1] This chemical reaction was one of the first homogeneous catalysis with organopalladium chemistry applied on an industrial scale.[2]
History[edit]
The Wacker reaction was first reported by Smidt et al.[3][4][5]
The development of the chemical process now known as the Wacker process began in 1956 at Wacker Chemie.[6] At the time, many industrial compounds were produced from acetylene, derived from calcium carbide, an expensive and environmentally unfriendly technology. The construction of a new oil refinery in Cologne by Esso close to a Wacker site, combined with the realization that ethylene would be a cheaper raw-material prompted Wacker to investigate its potential uses. As part of the ensuing research effort, a reaction of ethylene and oxygen over palladium on carbon in a quest for ethylene oxide unexpectedly gave evidence for the formation of acetaldehyde (simply based on smell). More research into this ethylene to acetaldehyde conversion resulted in a 1957 patent describing a gas-phase reaction using a heterogeneous catalyst.[7] In the meanwhile Hoechst AG joined the race and after a patent filing forced Wacker into a partnership called Aldehyd GmbH. The heterogeneous process ultimately failed due to catalyst inactivation and was replaced by the water-based homogeneous system for which a pilot plant was operational in 1958. Problems with the aggressive catalyst solution were solved by adopting titanium (newly available for industrial use) as construction material for reactors and pumps. Production plants went into operation in 1960.
Reaction mechanism[edit]
The reaction mechanism for the industrial Wacker process (olefin oxidation via palladium(II) chloride) has received significant attention for several decades. Aspects of the mechanism are still debated. A modern formulation is described below:
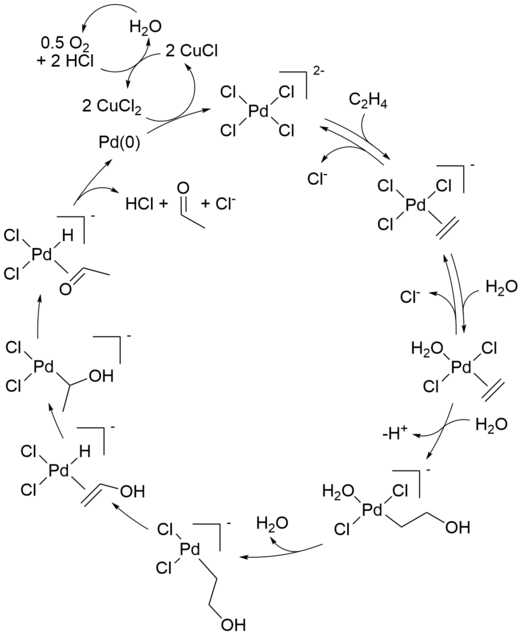
The initial stoichiometric reaction was first reported by Phillips.[9][10] The net reaction can also be described as follows:
- [PdCl4]2 − + C2H4 + H2O → CH3CHO + Pd + 2 HCl + 2 Cl−
This conversion is followed by reactions that regenerate the Pd(II) catalyst:
- Pd + 2 CuCl2 + 2 Cl − → [PdCl4]2− + 2 CuCl
- 2 CuCl + 1/2 O2 + 2 HCl → 2 CuCl2 + H2O
Only the alkene and oxygen are consumed. Without copper(II) chloride as an oxidizing agent, Pd(0) metal (resulting from beta-hydride elimination of Pd(II) in the final step) would precipitate, stopping the reaction after one cycle. This stoichiometric reaction was discovered in 1894. Air, pure oxygen, or a number of other reagents can then oxidise the resultant CuCl-chloride mixture back to CuCl2, allowing the cycle to continue.
Historical mechanistic studies[edit]
Early mechanistic studies from the 1960s elucidated several key points:[11][8]
- No H/D exchange effects seen in this reaction. Experiments using C2D4 in water generate CD3CDO, and runs with C2H4 in D2O generate CH3CHO. Thus, keto-enol tautomerization is not a possible mechanistic step.
- Negligible kinetic isotope effect with fully deuterated reactants (k H/k D=1.07). Hence, it is inferred that hydride transfer is not rate-determining.
- Significant competitive isotope effect with C2H2D2, (k H/k D= ~1.9), suggests that rate determining step be prior to formation of acetaldehyde.
- High concentrations of chloride and copper(II) chloride favor formation of a new product, chlorohydrin.
Many mechanistic studies on the Wacker process have focused on pathway for formation of the C-O bond, the hydroxypalladation step. Henry inferred that coordinated hydroxide attacks the ethylene ligand, an internal (syn-) pathway.[12] Later, stereochemical studies by Stille and coworkers[13][14][15] support an anti-addition pathway, whereby free hydroxide attacks the ethylene ligand. The conditions for Stille's experiments differ significantly from industrial process conditions. Other studies using normal industrial Wacker conditions (except with high chloride and high copper chloride concentrations) also yielded products that inferred nucleophilic attack was an anti-addition reaction.[16]
Kinetic studies were conducted on isotopically substituted allyl alcohols at standard industrial conditions (with low-chloride concentrations) to probe the reaction mechanisms.[17][18] Those results showed that nucleophilic attack is a slow process, while the proposed mechanisms explaining the earlier stereochemical studies assumed nucleophilic attack to be a fast process.
Subsequent stereochemical studies indicated that both pathways occur and are dependent on chloride concentrations.[19][20] However, these studies too are disputed since allyl-alcohols may be sensitive to isomerization reactions, and different stereoisomers may be formed from those reactions and not from the standard Wacker process.
In summary, experimental evidence seems to support that syn-addition occurs under low-chloride reaction concentrations (< 1 mol/L, industrial process conditions), while anti-addition occurs under high-chloride (> 3mol/L) reaction concentrations, probably due to chloride ions saturating the catalyst and inhibiting the inner-sphere mechanism. However, the exact pathway and the reason for this switching of pathways is still unknown.
Further complicating the Wacker process mechanism is questions about the role of copper chloride. Most theories assumed copper does not play a role in the olefin oxidation mechanisms. Yet, experiments by Stangl and Jira[21] found chlorohydrin formation was dependent on copper chloride concentrations. Work by Hosokawa and coworkers[22] yielded a crystallized product containing copper chloride, indicating it may have a non-innocent role in olefin oxidation. Finally, an ab initio study by Comas-Vives, et al. [23] involving no copper co-catalyst found anti-addition was the preferred pathway. This pathway was later confirmed by copper-free experiments by Anderson and Sigman.[24] A different kinetic rate law with no proton dependence was found under copper-free conditions, indicating the possibility that even small amounts of copper co-catalysts may have non-innocent roles on this chemistry. While these works complicate the picture of the Wacker process mechanism, one should probably infer that this and related chemistry can be sensitive to reaction conditions, and multiple different reaction pathways may be in play.
Another key step in the Wacker process is the migration of the hydrogen from oxygen to chloride and formation of the C-O double bond. This step is generally thought to proceed through a so-called β-hydride elimination with a cyclic four-membered transition state:

In silico studies[25][26][27] argue that the transition state for this reaction step is unfavorable and an alternative reductive elimination reaction mechanism is in play. The proposed reaction steps are likely assisted by water molecule in solution acting as a catalyst.

Industrial process[edit]
Two routes are commercialized for the production of acetaldehyde: one-stage process and two-stage.
One-stage process[edit]
Ethene and oxygen are passed co-currently in a reaction tower at about 130 °C and 400 kPa.[28] The catalyst is an aqueous solution of PdCl2 and CuCl2. The acetaldehyde is purified by extractive distillation followed by fractional distillation. Extractive distillation with water removes the lights ends having lower boiling points than acetaldehyde (chloromethane, chloroethane, and carbon dioxide) at the top, while water and higher-boiling byproducts, such as acetic acid, crotonaldehyde or chlorinated acetaldehydes, are withdrawn together with acetaldehyde at the bottom.[28] Due to the corrosive nature of catalyst, the reactor is lined with acid-proof ceramic material and the tubing is made of titanium.
Two-stage process[edit]
In two-stage process, reaction and oxidation are carried out separately in tubular reactors. Unlike one-stage process, air can be used instead of oxygen. Ethylene is passed through the reactor along with catalyst at 105–110 °C and 900–1000 kPa.[28] Catalyst solution containing acetaldehyde is separated by flash distillation. The catalyst is oxidized in the oxidation reactor at 1000 kPa using air as oxidizing medium. Oxidized catalyst solution is separated and sent back to reactor. Oxygen from air is used up completely and the exhaust air is circulated as inert gas. Acetaldehyde – water vapor mixture is preconcentrated to 60–90% acetaldehyde by utilizing the heat of reaction and the discharged water is returned to the flash tower to maintain catalyst concentration. A two-stage distillation of the crude acetaldehyde follows. In the first stage, low-boiling substances, such as chloromethane, chloroethane and carbon dioxide, are separated. In the second stage, water and higher-boiling by-products, such as chlorinated acetaldehydes and acetic acid, are removed and acetaldehyde is obtained in pure form overhead.[28] Due to corrosive nature of the catalyst, the equipments in contact with it are lined with titanium.
In both one- and two-stage processes the acetaldehyde yield is about 95%[28] and the production costs are virtually the same. The advantage of using dilute gases in the two-stage method is balanced by higher investment costs. Both methods yield chlorinated hydrocarbons, chlorinated acetaldehydes, and acetic acid as byproducts. Generally, the choice of method is governed by the raw material and energy situations as well as by the availability of oxygen at a reasonable price. In general, 100 parts of ethene gives:
- 95 parts acetaldehyde
- 1.9 parts chlorinated aldehydes
- 1.1 parts unconverted ethene
- 0.8 parts carbon dioxide
- 0.7 parts acetic acid
- 0.1 parts chloromethane
- 0.1 parts ethyl chloride
- 0.3 parts ethane, methane, crotonaldehyde
and other minor side products
-
 A flow chart showing the process flow diagram for the one-stage Wacker Process for manufacture of acetaldehyde.
A flow chart showing the process flow diagram for the one-stage Wacker Process for manufacture of acetaldehyde. -
 A flow chart showing the process flow diagram for the two-stage Wacker process for manufacture of acetaldehyde.
A flow chart showing the process flow diagram for the two-stage Wacker process for manufacture of acetaldehyde.


Tsuji-Wacker oxidation[edit]
The advent of Wacker Process has spurred on many investigations into the utility and applicability of the reactions to more complex terminal olefins. The Tsuji-Wacker oxidation is the palladium(II)-catalyzed transformation of such olefins into carbonyl compounds. Clement and Selwitz[29] were the first to find that using an aqueous DMF as solvent allowed for the oxidation of 1-dodecene to 2-dodecanone, which addressed the insolubility problem of higher order olefins in water. Fahey[30] noted the use of 3-methylsulfolane in place of DMF as solvent increased the yield of oxidation of 3,3-Dimethylbut-1-ene. Two years after, Tsuji[31] applied the Selwitz conditions for selective oxidations of terminal olefins with multiple functional groups, and demonstrated its utility in synthesis of complex substrates.[32] Further development of the reaction has led to various catalytic systems to address selectivity of the reaction, as well as introduction of intermolecular and intramolecular oxidations with non-water nucleophiles.
Regioselectivity[edit]
Markovnikov addition[edit]
The Tsuji-Wacker oxidation oxidizes terminal olefin to the corresponding methyl ketone under the Wacker process condition. Almost identical to that of Wacker Process, the proposed catalytic cycle[33](Figure 1) begins with complexation of PdCl2 and two chloride anions to PdCl4, which then undergoes subsequent ligand exchange of two chloride ligand for water and alkene to form Pd(Cl2)(H2O)(alkene) complex. A water molecule then attacks the olefin regioselectively through an outer sphere mechanism in a Markovnikov fashion, to form the more thermodynamically stable Pd(Cl2)(OH)(-CH2-CHOH-R) complex. Dissociation of a chloride ligand to the three coordinate palladium complex promotes β-hydride elimination, then subsequent 1,2-hydride migratory insertion generates Pd(Cl2)(OH)(-CHOHR-CH3) complex. This undergoes β-hydride elimination to release the ketone, and subsequent reductive elimination produces HCl, water, and palladium(0). Finally palladium(0) is reoxidized to PdCl2 with two equivalents of Cu(II)Cl2, which in turn can be reoxidized by O2.
The oxidation of terminal olefins generally provide the Markovnikov ketone product, however in cases where substrate favors the aldehyde (discussed below), different ligands can be used to enforce the Markovnikov regioselectivity. The use of sparteine as a ligand (Figure 2, A)[34] favors nucleopalladation at the terminal carbon to minimize steric interaction between the palladium complex and substrate. The Quinox-ligated palladium catalyst is used to favor ketone formation when substrate contains a directing group (Figure 2, B).[35] When such substrate bind to Pd(Quinox)(OOtBu), this complex is coordinately saturated which prevents the binding of the directing group, and results in formation of the Markovnikov product. The efficiency of this ligand is also attributed to its electronic property, where anionic TBHP prefers to bind trans to the oxazoline and olefin coordinate trans to the quinoline.[36]

Anti-Markovnikov addition [edit]
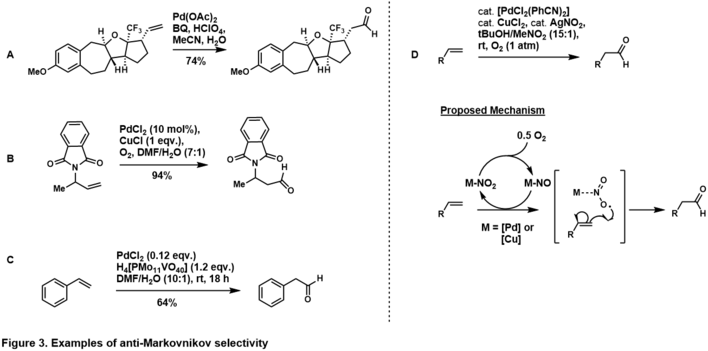
The anti-Markovnikov addition selectivity to aldehyde can be achieved through exploiting inherent stereoelectronics of the substrate.[37] Placement of directing group at homo-allylic (i.e. Figure 3, A)[38] and allylic position (i.e. Figure 3, B)[39] to the terminal olefin favors the anti-Markovnikov aldehyde product, which suggests that in the catalytic cycle the directing group chelates to the palladium complex such that water attacks at the anti-Markovnikov carbon to generate the more thermodynamically stable palladacycle. Anti-Markovnikov selectivity is also observed in styrenyl substrates (i.e. Figure 3, C),[40] presumably via η4-palladium-styrene complex after water attacks anti-Markovnikov. More examples of substrate-controlled, anti-Markovnikov Tsuji-Wacker Oxidation of olefins are given in reviews by Namboothiri,[41] Feringa,[37] and Muzart.[42]
Grubbs and co-workers paved way for anti-Markovnikov oxidation of stereoelectronically unbiased terminal olefins, through the use of palladium-nitrite system (Figure 2, D).[43] In his system, the terminal olefin was oxidized to the aldehyde with high selectivity through a catalyst-control pathway. The mechanism is under investigation, however evidence[41] suggests it goes through a nitrite radical adds into the terminal carbon to generate the more thermodynamically stable, secondary radical. Grubbs expanded this methodology to more complex, unbiased olefins.[44][45]
Scope[edit]
Oxygen nucleophiles [edit]
The intermolecular oxidations of olefins with alcohols as nucleophile typically generate ketals, where as the palladium-catalyzed oxidations of olefins with carboxylic acids as nucleophile generates vinylic or allylic carboxylates. In case of diols, their reactions with alkenes typically generate ketals, whereas reactions of olefins bearing electron-withdrawing groups tend to form acetals.[46]
Palladium-catalyzed intermolecular oxidations of dienes with carboxylic acids and alcohols as donors give 1,4-addition products. In the case of cyclohexadiene (Figure 4, A), Backvall found that stereochemical outcome of product was found to depend on concentration of LiCl.[47] This reaction proceeds by first generating the Pd(OAc)(benzoquinone)(allyl) complex, through anti-nucleopalladation of diene with acetate as nucleophile. The absence of LiCl induces an inner sphere reductive elimination to afford the trans-acetate stereochemistry to give the trans-1,4-adduct. The presence of LiCl displaces acetate with chloride due to its higher binding affinity, which forces an outer sphere acetate attack anti to the palladium, and affords the cis-acetate stereochemistry to give the cis-1,4-adduct. Intramolecular oxidative cyclization: 2-(2-cyclohexenyl)phenol cyclizes to corresponding dihydro-benzofuran (Figure 4, B);[48] 1-cyclohexadiene-acetic acid in presence of acetic acid cyclizes to corresponding lactone-acetate 1,4 adduct (Figure 4, C),[49] with cis and trans selectivity controlled by LiCl presence.

Nitrogen nucleophiles[edit]
The oxidative aminations of olefins are generally conducted with amides or imides; amines are thought to be protonated by the acidic medium or to bind the metal center too tightly to allow for the catalytic chemistry to occur.[46] These nitrogen nucleophiles are found to be competent in both intermolecular and intramolecular reactions, some examples are depicted (Figure 5, A,[50] B[51])

References[edit]
- ^ Translated in part from de:Wacker-Verfahren.
- ^ Elschenbroich, C. "Organometallics" (2006) Wiley-VCH: Weinheim. ISBN 978-3-527-29390-2
- ^ J. Smidt, W. Hafner, R. Jira, J. Sedlmeier, R. Sieber, R. Rüttinger, and H. Kojer, Angew. Chem., 1959, 71, 176–182. doi:10.1002/ange.19590710503
- ^ W. Hafner, R. Jira, J. Sedlmeier, and J. Smidt, Chem. Ber., 1962, 95, 1575–1581.
- ^ J. Smidt, W. Hafner, R. Jira, R. Sieber, J. Sedlmeier, and A. Sabel, Angew. Chem. Int. Ed. Engl., 1962, 1, 80–88.
- ^ Acetaldehyde from Ethylene — A Retrospective on the Discovery of the Wacker Process Reinhard Jira Angew. Chem. Int. Ed. 2009, 48, 9034–9037 doi:10.1002/anie.200903992
- ^ J. Smidt, W. Hafner, J. Sedlmeier, R. Jira, R. Rottinger (Cons. f.elektrochem.Ind.), DE 1 049 845, 1959, Anm. 04.01.1957.
- ^ a b J. A. Keith; P. M. Henry (2009). "The Mechanism of the Wacker Reaction: A Tale of Two Hydroxypalladations". Angew. Chem. Int. Ed. 48 (48): 9038–9049. doi:10.1002/anie.200902194. PMID 19834921.
- ^ F. C. Phillips, Am. Chem. J., 1894, 16, 255–277.
- ^ F. C. Phillips, Z. Anorg. Chem., 1894, 6, 213–228.
- ^ Henry, Patrick M. In Handbook of Organopalladium Chemistry for Organic Synthesis; Negishi, E., Ed.; Wiley & Sons: New York, 2002; p 2119. ISBN 0-471-31506-0
- ^ P. M. Henry, J. Am. Chem. Soc., 1964, 86, 3246–3250.
- ^ James, D.E., Stille, J.K. J. Organomet. Chem., 1976, 108, 401. doi:10.1021/ja00423a028
- ^ Stille, J.K., Divakarumi, R.J., J. Organomet. Chem., 1979, 169, 239;
- ^ James, D.E., Hines, L.F., Stille, J.K. J. Am. Chem. Soc., 1976, 98, 1806 doi:10.1021/ja00423a027
- ^ Bäckvall, J.E., Akermark, B., Ljunggren, S.O., J. Am. Chem. Soc., 1979, 101, 2411. doi:10.1021/ja00503a029
- ^ Zaw, K., Lautens, M. and Henry P.M. Organometallics, 1985, 4, 1286–1296
- ^ Wan W.K., Zaw K., and Henry P.M. Organometallics, 1988, 7, 1677–1683
- ^ Francis, J.W., Henry, P.M. Organometallics, 1991, 10, 3498. doi:10.1021/om00056a019
- ^ Francis, J.W., Henry, P.M. Organometallics, 1992, 11, 2832.doi:10.1021/om00044a024
- ^ H. Stangl and R. Jira, Tetrahedron Lett., 1970, 11, 3589–3592
- ^ T. Hosokawa, T. Nomura, S.-I. Murahashi, J. Organomet. Chem., 1998, 551, 387–389
- ^ Comas-Vives, A., Stirling, A., Ujaque, G., Lledós, A., Chem. Eur. J., 2010, 16, 8738–8747.doi:10.1002/chem.200903522
- ^ Anderson, B.J., Keith, J.A., and Sigman, M.S., J. Am. Chem. Soc., 2010, 132, 11872-11874
- ^ J. A. Keith, J. Oxgaard, and W. A. Goddard, III J. Am. Chem. Soc., 2006, 128, 3132 – 3133; doi:10.1021/ja0533139
- ^ H. E. Hosseini, S. A. Beyramabadi, A. Morsali, and M. R. Housaindokht, J. Mol. Struct. (THEOCHEM), 2010, 941, 138–143
- ^ P. L. Theofanis, and W. A. Goddard, III Organometallics, 2011, 30, 4941 – 4948; doi:10.1021/om200542w
- ^ a b c d e Marc Eckert; Gerald Fleischmann; Reinhard Jira; Hermann M. Bolt; Klaus Golka. "Acetaldehyde". Ullmann's Encyclopedia of Industrial Chemistry. Weinheim: Wiley-VCH. doi:10.1002/14356007.a01_031.pub2. ISBN 978-3527306732.
- ^ Clement, William H.; Selwitz, Charles M. (January 1964). "Improved Procedures for Converting Higher α-Olefins to Methyl Ketones with Palladium Chloride". The Journal of Organic Chemistry. 29 (1): 241–243. doi:10.1021/jo01024a517. ISSN 0022-3263.
- ^ Fahey, Darryl R.; Zeuch, Ernest A. (November 1974). "Aqueous sulfolane as solvent for rapid oxidation of higher .alpha.-olefins to ketones using palladium chloride". The Journal of Organic Chemistry. 39 (22): 3276–3277. doi:10.1021/jo00936a023. ISSN 0022-3263.
- ^ Tsuji, Jiro; Shimizu, Isao; Yamamoto, Keiji (August 1976). "Convenient general synthetic method for 1,4- and 1,5-diketones by palladium catalyzed oxidation of α-allyl and α-3-butenyl ketones". Tetrahedron Letters. 17 (34): 2975–2976. doi:10.1016/s0040-4039(01)85504-0. ISSN 0040-4039.
- ^ Tsuji, Jiro (1984). "Synthetic Applications of the Palladium-Catalyzed Oxidation of Olefins to Ketones". Synthesis. 1984 (5): 369–384. doi:10.1055/s-1984-30848. ISSN 0039-7881. S2CID 95604861.
- ^ Kurti, Laszlo; Czako, Barbara (2005). Strategic Applications of named Reactions in Organic Synthesis. 525 B Street, Suite 1900, San Diego, California 92101-4495, USA: Elsevier Academic Press. p. 474. ISBN 978-0-12-429785-2.
{{cite book}}: CS1 maint: location (link) - ^ Balija, Amy M.; Stowers, Kara J.; Schultz, Mitchell J.; Sigman, Matthew S. (March 2006). "Pd(II)-Catalyzed Conversion of Styrene Derivatives to Acetals: Impact of (−)-Sparteine on Regioselectivity". Organic Letters. 8 (6): 1121–1124. doi:10.1021/ol053110p. ISSN 1523-7060. PMID 16524283.
- ^ Michel, Brian W.; Camelio, Andrew M.; Cornell, Candace N.; Sigman, Matthew S. (2009-05-06). "A General and Efficient Catalyst System for a Wacker-Type Oxidation Using TBHP as the Terminal Oxidant: Application to Classically Challenging Substrates". Journal of the American Chemical Society. 131 (17): 6076–6077. doi:10.1021/ja901212h. ISSN 0002-7863. PMC 2763354. PMID 19364100.
- ^ Michel, Brian W.; Steffens, Laura D.; Sigman, Matthew S. (June 2011). "On the Mechanism of the Palladium-Catalyzed tert -Butylhydroperoxide-Mediated Wacker-Type Oxidation of Alkenes Using Quinoline-2-Oxazoline Ligands". Journal of the American Chemical Society. 133 (21): 8317–8325. doi:10.1021/ja2017043. ISSN 0002-7863. PMC 3113657. PMID 21553838.
- ^ a b Dong, Jia Jia; Browne, Wesley R.; Feringa, Ben L. (2014-11-03). "Palladium-Catalyzed anti-Markovnikov Oxidation of Terminal Alkenes" (PDF). Angewandte Chemie International Edition. 54 (3): 734–744. doi:10.1002/anie.201404856. ISSN 1433-7851. PMID 25367376.
- ^ Miller, D. G.; Wayner, Danial D. M. (April 1990). "Improved method for the Wacker oxidation of cyclic and internal olefins". The Journal of Organic Chemistry. 55 (9): 2924–2927. doi:10.1021/jo00296a067. ISSN 0022-3263.
- ^ Stragies, Roland; Blechert, Siegfried (October 2000). "Enantioselective Synthesis of Tetraponerines by Pd- and Ru-Catalyzed Domino Reactions". Journal of the American Chemical Society. 122 (40): 9584–9591. doi:10.1021/ja001688i. ISSN 0002-7863.
- ^ Wright, Joseph A.; Gaunt, Matthew J.; Spencer, Jonathan B. (2006-01-11). "Novel Anti-Markovnikov Regioselectivity in the Wacker Reaction of Styrenes". Chemistry - A European Journal. 12 (3): 949–955. doi:10.1002/chem.200400644. ISSN 0947-6539. PMID 16144020.
- ^ a b Baiju, Thekke Veettil; Gravel, Edmond; Doris, Eric; Namboothiri, Irishi N.N. (September 2016). "Recent developments in Tsuji-Wacker oxidation". Tetrahedron Letters. 57 (36): 3993–4000. doi:10.1016/j.tetlet.2016.07.081. ISSN 0040-4039.
- ^ Muzart, Jacques (August 2007). "Aldehydes from Pd-catalysed oxidation of terminal olefins". Tetrahedron. 63 (32): 7505–7521. doi:10.1016/j.tet.2007.04.001. ISSN 0040-4020.
- ^ Wickens, Zachary K.; Morandi, Bill; Grubbs, Robert H. (2013-09-13). "Aldehyde-Selective Wacker-Type Oxidation of Unbiased Alkenes Enabled by a Nitrite Co-Catalyst" (PDF). Angewandte Chemie International Edition. 52 (43): 11257–11260. doi:10.1002/anie.201306756. ISSN 1433-7851. PMID 24039135.
- ^ Wickens, Zachary K.; Skakuj, Kacper; Morandi, Bill; Grubbs, Robert H. (2014-01-13). "Catalyst-Controlled Wacker-Type Oxidation: Facile Access to Functionalized Aldehydes" (PDF). Journal of the American Chemical Society. 136 (3): 890–893. doi:10.1021/ja411749k. ISSN 0002-7863. PMID 24410719.
- ^ Kim, Kelly E.; Li, Jiaming; Grubbs, Robert H.; Stoltz, Brian M. (2016-09-30). "Catalytic Anti-Markovnikov Transformations of Hindered Terminal Alkenes Enabled by Aldehyde-Selective Wacker-Type Oxidation" (PDF). Journal of the American Chemical Society. 138 (40): 13179–13182. doi:10.1021/jacs.6b08788. ISSN 0002-7863. PMID 27670712.
- ^ a b Hartwig, John F. (2010). Organotransition Metal Chemistry: From Bonding to Catalysis. USA: University Science Books. pp. 717–734. ISBN 978-1-891389-53-5.
- ^ Baeckvall, Jan E.; Bystroem, Styrbjoern E.; Nordberg, Ruth E. (November 1984). "Stereo- and regioselective palladium-catalyzed 1,4-diacetoxylation of 1,3-dienes". The Journal of Organic Chemistry. 49 (24): 4619–4631. doi:10.1021/jo00198a010. ISSN 0022-3263.
- ^ Hosokawa, Takahiro; Miyagi, Shyogo; Murahashi, Shunichi; Sonoda, Akio (July 1978). "Oxidative cyclization of 2-allylphenols by palladium(II) acetate. Changes in product distribution". The Journal of Organic Chemistry. 43 (14): 2752–2757. doi:10.1021/jo00408a004. ISSN 0022-3263.
- ^ Baeckvall, Jan E.; Granberg, Kenneth L.; Andersson, Pher G.; Gatti, Roberto; Gogoll, Adolf (September 1993). "Stereocontrolled lactonization reactions via palladium-catalyzed 1,4-addition to conjugated dienes". The Journal of Organic Chemistry. 58 (20): 5445–5451. doi:10.1021/jo00072a029. ISSN 0022-3263.
- ^ Timokhin, Vitaliy I.; Stahl, Shannon S. (December 2005). "Brønsted Base-Modulated Regioselectivity in the Aerobic Oxidative Amination of Styrene Catalyzed by Palladium". Journal of the American Chemical Society. 127 (50): 17888–17893. doi:10.1021/ja0562806. ISSN 0002-7863. PMID 16351120.
- ^ Larock, Richard C.; Hightower, Timothy R.; Hasvold, Lisa A.; Peterson, Karl P. (January 1996). "Palladium(II)-Catalyzed Cyclization of Olefinic Tosylamides". The Journal of Organic Chemistry. 61 (11): 3584–3585. doi:10.1021/jo952088i. ISSN 0022-3263. PMID 11667199.
| Authority control databases: National |
|---|