
Retinoblastoma
| Retinoblastoma | |
|---|---|
 | |
| A pathology specimen of a retinoblastoma tumor from an enucleated eye of a 3-year-old female | |
| Specialty | Neuro-oncology |
| Symptoms | Leukocoria seen in patient's pupil in photos Poor vision One or both eyes turning inward or outward Eye pain[1] |
| Usual onset | Under 3 years old[1] |
| Treatment | Surgery (including eye removal in advanced cases) Chemotherapy (after surgery in cases of metastasis) Focal therapy[1] |
| Frequency | ~250–300 children diagnosed annually (United States)[1] |
Retinoblastoma (Rb) is a rare form of cancer that rapidly develops from the immature cells of a retina,[2] the light-detecting tissue of the eye.[3] It is the most common primary malignant intraocular cancer in children, and it is almost exclusively found in young children.[4]
Though most children in high income countries survive this cancer,[2] they may lose their vision in the affected eye(s)[5] or need to have the eye removed.[2]
Almost half of children with retinoblastoma have a hereditary genetic defect associated with retinoblastoma. In other cases, it is caused by a congenital mutation in the chromosome 13 gene 13q14 (retinoblastoma protein).[6]
Signs and symptoms[edit]


Retinoblastoma is the most intrusive intraocular cancer among children. The chance of survival and preservation of the eye depends fully on the severity. Retinoblastoma is extremely rare as there are only about 200 to 300 cases every year in the United States. Globally, only 1 in about 15,000 children have this malignancy, though rates continue to increase.[3]
Intraocular malignancies are relatively more frequently treated than extraocular malignancies, likely due to a relatively earlier detection and subsequent treatment. Pediatricians may screen infants with annual vision tests, in which anomalies can be detected. During a red reflex test, light from an ophthalmoscope goes through transparent parts of the eye and reflects off the ocular fundus. If retinoblastoma is present, it may partially or fully impede light transversing this path. This may result in an abnormal red reflex or leucocoria, which can be a common indicator of retinoblastoma (when light is reflected by the tumor, the regular view of the red retina is blocked). The retinoblastoma may be visible as a whitish, translucent mass. If the tumor has not spread and is contained within the eye, chances of successful treatment are favorable. If initial signs are ignored or diagnosis is significantly delayed, outcomes and prognosis worsen. The effects of retinoblastoma may spread outside the eye, sometimes resulting in proptosis. Retinoblastoma that has spread may be significantly more difficult to treat. [7]
The most common and obvious sign of retinoblastoma is an abnormal appearance of the retina as viewed through the pupil, the medical term for which is leukocoria, also known as amaurotic cat's eye reflex.[4] Other signs and symptoms include deterioration of vision, a red and irritated eye with glaucoma, and faltering growth or delayed development. Some children with retinoblastoma can develop a squint,[8] commonly referred to as "cross-eyed" or "wall-eyed" (strabismus). Retinoblastoma presents with advanced disease in developing countries and eye enlargement is a common finding.[9]
Depending on the position of the tumors, they may be visible during a simple eye examination using an ophthalmoscope to look through the pupil. A positive diagnosis is usually made only with an examination under anesthetic (EUA). A white eye reflection is not always a positive indication of retinoblastoma and can be caused by light being reflected badly[10] or by other conditions such as Coats' disease.[11]
The presence of the photographic fault red eye in only one eye and not in the other may be a sign of retinoblastoma. A clearer sign is "white eye" or "cat's eye" (leukocoria).[12]
Cause[edit]
Mutation of genes, found in chromosomes, can affect the way in which cells grow and develop within the body.[13] Alterations in RB1 or MYCN can give rise to retinoblastoma.
RB1[edit]
In children with the heritable genetic form of retinoblastoma, a mutation occurs in the RB1 gene on chromosome 13. RB1 was the first tumor suppressor gene cloned.[13] Although RB1 interacts with over 100 cell proteins,[13] its negative regulator effect on the cell cycle principally arises from binding and inactivation of the transcription factor E2F, thus repressing the transcription of genes which are required for the S phase.[13]
The defective RB1 gene can be inherited from either parent; in some children, however, the mutation occurs in the early stages of fetal development. The expression of the RB1 allele is autosomal dominant with 90% penetrance.[14]
Inherited forms of retinoblastomas are more likely to be bilateral. In addition, inherited uni- or bilateral retinoblastomas may be associated with pineoblastoma and other malignant midline supratentorial primitive neuroectodermal tumors (PNETs) with a dismal outcome; retinoblastoma concurrent with a PNET is known as trilateral retinoblastoma.[15] A 2014 meta-analysis showed that 5-year survival of trilateral retinoblastoma increased from 6% before 1995 to 57% by 2014, attributed to early detection and improved chemotherapy.[16]
The development of retinoblastoma can be explained by the two-hit model. According to the two-hit model, both alleles need to be affected, so two events are necessary for the retinal cell or cells to develop into tumors. The first mutational event can be inherited (germline or constitutional), which will then be present in all cells in the body. The second “hit” results in the loss of the remaining normal allele (gene) and occurs within a particular retinal cell.[17] In the sporadic, nonheritable form of retinoblastoma, both mutational events occur within a single retinal cell after fertilization (somatic events); sporadic retinoblastoma tends to be unilateral.
Several methods have been developed to detect the RB1 gene mutations.[18][19] Attempts to correlate gene mutations to the stage at presentation have not shown convincing evidence of a correlation.[20]
MYCN[edit]
Not all retinoblastoma cases are with RB1 inactivation. There are cases reported with only one RB1 mutation or even two functional RB1 alleles, which indicates other oncogenic lesions of retinoblastoma.[21] Somatic amplification of the MYCN oncogene is responsible for some cases of nonhereditary, early-onset, aggressive, unilateral retinoblastoma. MYCN can act as a transcription factor and promotes proliferation by regulating the expression of cell cycle genes.[22][23] Although MYCN amplification accounted for only 1.4% of retinoblastoma cases, researchers identified it in 18% of infants diagnosed at less than 6 months of age. Median age at diagnosis for MYCN retinoblastoma was 4.5 months, compared with 24 months for those who had nonfamilial unilateral disease with two RB1 gene mutations.[24]
Diagnosis[edit]

Screening for retinoblastoma should be part of a "well baby" screening for newborns during the first 3 months of life, to include:[25]
- The red reflex: checking for a normal reddish-orange reflection from the eye's retina with an ophthalmoscope or retinoscope from about 30 cm or 1 foot, usually done in a dimly lit or dark room
- The corneal light reflex or Hirschberg test: checking for symmetrical reflection of beam of light in the same spot on each eye when a light is shined into each cornea, to help determine whether the eyes are crossed
- Eye examination: checking for any structural abnormalities
Classification[edit]
The two forms of the disease are a heritable form and nonheritable form (all cancers are considered genetic in that mutations of the genome are required for their development, but this does not imply that they are heritable, or transmitted to offspring). Approximately 40% of patients have a heritable form of retinoblastoma, carrying a mutation in the RB1 gene.[26] If no history of the disease exists within the family, the disease is labeled "sporadic", but this does not necessarily indicate that it is the nonheritable form. Bilateral retinoblastomas are commonly heritable, while unilateral retinoblastomas are commonly nonheritable.[citation needed]
In about two-thirds of cases,[27] only one eye is affected (unilateral retinoblastoma); in the other third, tumors develop in both eyes (bilateral retinoblastoma). The number and size of tumors on each eye may vary. In certain cases, the pineal gland or the suprasellar or parasellar region (or in very rare cases other midline intracranial locations) is also affected (trilateral retinoblastoma). The position, size, and quantity of tumors are considered when choosing the type of treatment for the disease.[citation needed]
Differential diagnosis[edit]

- 1. Persistent fetal vasculature is a congenital developmental anomaly of the eye resulting from failure of the embryological, primary vitreous, and hyaloid vasculature to regress, whereby the eye is shorter, develops a cataract, and may present with whitening of the pupil.
- 2. Coats disease is a typically unilateral disease characterised by abnormal development of blood vessels behind the retina, leading to blood vessel abnormalities in the retina and retinal detachment to mimic retinoblastoma.
- 3. Toxocariasis is a parasitic disease of the eye associated with exposure to infected puppies, which causes a retinal lesion leading to retinal detachment.
- 4. Retinopathy of prematurity is associated with low-birth-weight infants who receive supplemental oxygen in the period immediately after birth, and it involves damage to the retinal tissue and may lead to retinal detachment.
- 5. Congenital Cataract
- 6. Norrie Disease
If the eye examination is abnormal, further testing may include imaging studies, such as computerized tomography (CT), magnetic resonance imaging (MRI), and ultrasound.[28] CT and MRI can help define the structure abnormalities and reveal any calcium depositions. Ultrasound can help define the height and thickness of the tumor. Bone marrow examination or lumbar puncture may also be done to determine any metastases to bones or the brain.[citation needed]
Morphology[edit]
Gross and microscopic appearances of retinoblastoma are identical in both hereditary and sporadic types. Macroscopically, viable tumor cells are found near blood vessels, while zones of necrosis are found in relatively avascular areas. Microscopically, both undifferentiated and differentiated elements may be present. Undifferentiated elements appear as collections of small, round cells with hyperchromatic nuclei; differentiated elements include Flexner-Wintersteiner rosettes, Homer Wright rosettes,[29] and fleurettes from photoreceptor differentiation.[30]
-
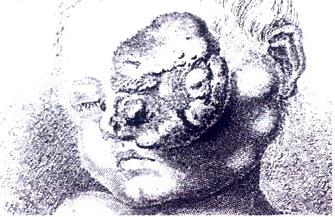 Drawing of a large retinoblastoma
Drawing of a large retinoblastoma -
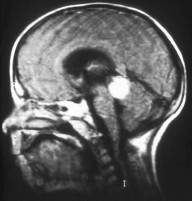 Aspect of trilateral retinoblastoma on MRI
Aspect of trilateral retinoblastoma on MRI -
 An ocular ultrasound of a large retinoblastoma tumor within the eye of a 3-year-old boy
An ocular ultrasound of a large retinoblastoma tumor within the eye of a 3-year-old boy -
 Funduscopic finding of a retinoblastoma
Funduscopic finding of a retinoblastoma -
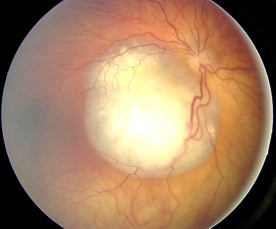 Ocular fundus aspect of retinoblastoma
Ocular fundus aspect of retinoblastoma -
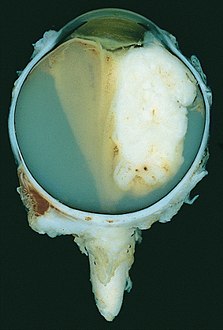 Large exophytic white tumor with foci of calcification producing total exudative retinal detachment
Large exophytic white tumor with foci of calcification producing total exudative retinal detachment -
 Flexner-Wintersteiner rosettes in retinoblastoma
Flexner-Wintersteiner rosettes in retinoblastoma -
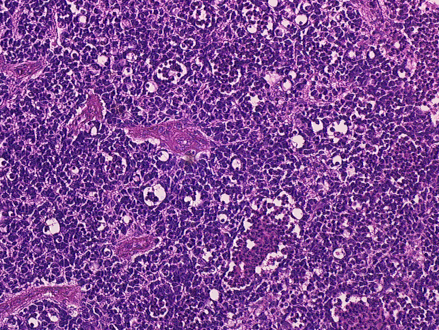 Retinoblastoma, 400 X magnification
Retinoblastoma, 400 X magnification -
 Crystal structure of the retinoblastoma tumor suppressor protein bound to E2F peptide polymer
Crystal structure of the retinoblastoma tumor suppressor protein bound to E2F peptide polymer









Genetic testing[edit]
Identifying the RB1 gene mutation that led to a child's retinoblastoma can be important in the clinical care of the affected individual and in the care of (future) siblings and offspring. It may run in the family.
- Bilaterally affected individuals and 13-15% of unilaterally affected individuals,[31][32] are expected to show an RB1 mutation in blood. By identifying the RB1 mutation in the affected individual, (future) siblings, children, and other relatives can be tested for the mutation; if they do not carry the mutation, child relatives are not at risk of retinoblastoma, so need not undergo the trauma and expense of examinations under anaesthetic.[33] For the 85% of unilaterally affected patients found not to carry either of their eye tumor RB1 mutations in blood, neither molecular testing nor clinical surveillance of siblings is required.
- If the RB1 mutation of an affected individual is identified, amniotic cells in an at-risk pregnancy can be tested for the family mutation; any fetus that carries the mutation can be delivered early, allowing early treatment of any eye tumors, leading to better visual outcomes.[33]
- For cases of unilateral retinoblastoma where no eye tumor is available for testing, if no RB1 mutation is detected in blood after high-sensitivity molecular testing (i.e. >93% RB1 mutation detection sensitivity), the risk of a germline RB1 mutation is reduced to less than 1%,[32] a level at which only clinic examination (and not examinations under anaesthetic) is recommended for the affected individual and their future offspring (National Retinoblastoma Strategy, Canadian Guidelines for Care).[34]
Imaging[edit]
Traditional ultrasound B scan can detect calcifications in the tumour while high-frequency ultrasound B scan is able to provide higher resolution than the traditional ultrasound and determine the proximity of the tumour with front portion of the eye. MRI scan can detect high-risk features such as optic nerve invasion; choroidal invasion, scleral invasion, and intracranial invasion. CT scan is generally avoided because radiation can stimulate the formation of more eye tumours in those with RB1 genetic mutation.[35]
Staging[edit]
In order to properly diagnose retinoblastoma, there must be guidelines to follow to properly classify the risk of the tumor. The Reese Ellsworth Classification System, by Dr. Algernon Reese and Dr. Robert Ellsworth, is universally used to determine the size, location, and multi-focality of the tumor.[36] The system was originally used to decide the best treatment result by using external beam radiotherapy, as well as, the likeliness of salvaging the globe of the eye. Due to chemotherapy not being part of the Reese Ellsworth Classification System, there needed to be an updated classification system to foresee the treatment outcomes of chemotherapy. The International Classification for Intraocular Retinoblastoma is now the current system being used, and it was created by Murphree and associates.[36] According to Reese and Ellsworth, there were different groups that had various features in order to classify the globe salvage as very favorable to the category of very unfavorable. In order to salvage the affected eye, the disc diameter had to be around 4DD and behind the equator to have higher favorability. If the tumor was around ten in disc diameter and involved roughly 50% of the retina, it was considered unfavorable to salvage the globe which could result in enucleation. According to Murphree, the different groups were classified from very low risk to very high risk which was determined by features of the given tumor. Very low risk means that the tumor has to be less than 3mm and there must be no seeding of the vitreous or sub-retinal area. When a patient is very high risk, the tumor presents itself with multiple features and is going to have to be treated with conservative treatment modalities or enucleation.[37]
| Group | Clinical Features |
| A
Very low risk |
All tumors are 3mm or smaller, confined to the retina, and located at least 3mm from the foveola and 1.5mm from the optic nerve. No vitreous or subretinal seeding |
| B
Low risk |
Retinal tumors may be any size or location not in Group A, No vitreous or subretinal seeding allowed. A small cuff of subretinal fluid extending no more than 5mm from the base of the tumor is allowed |
| C
Moderate risk |
Eyes with only focal vitreous or subretinal seeding and discrete retinal tumors of any size and location. Vitreous or subretinal seeding may extend no more than 3mm from the tumor. Up to one quadrant of subretinal fluid may be present |
| D
High risk |
Eyes with diffuse vitreous or subretinal seeding and/or massive, nondiscrete endophytic or exophytic disease. More than one quadrant of retinal detachment |
| E
Very high risk eyes |
Eyes with one or more of the following:
Irreversible neovascular glaucoma Massive intraocular hemorrhage Aseptic orbital cellulitis Phthisis or pre-phthisis Tumor anterior to anterior vitreous face Tumor touching the lens Diffuse infiltrating retinoblastoma |
International Classification for Intraocular Retinoblastoma[36][37]
Treatment[edit]

The priority of retinoblastoma treatment is to preserve the life of the child, then to preserve vision, and then to minimize complications or side effects of treatment. The exact course of treatment depends on the individual case and is decided by the ophthalmologist in discussion with the paediatric oncologist.[38] Correct treatment also depends on the mutation type, whether it is a germline RB1 mutation, a sporadic RB1 mutation or MYCN amplification with functional RB1.[39] Children with involvement of both eyes at diagnosis usually require multimodality therapy (chemotherapy, local therapies).
The various treatment modalities for retinoblastoma includes:[38]
- Enucleation of the eye – Most patients with unilateral disease present with advanced intraocular disease, so usually undergo enucleation, which results in a cure rate of 95%. In bilateral Rb, enucleation is usually reserved for eyes that have failed all known effective therapies or without useful vision.
- External beam radiotherapy (EBRT) – The most common indication for EBRT is for the eye in a young child with bilateral retinoblastoma who has active or recurrent disease after completion of chemotherapy and local therapies. However, patients with hereditary disease who received EBRT therapy are reported to have a 35% risk of second cancers.[40]
- Brachytherapy involves the placement of a radioactive implant (plaque), usually on the sclera adjacent to the base of a tumor. It used as the primary treatment, or more frequently, in patients with small tumors or in those who had failed initial therapy including previous EBRT therapy.
- Thermotherapy involves the application of heat directly to the tumor, usually in the form of infrared radiation. It is also used for small tumors.
- Laser photocoagulation is recommended only for small posterior tumors. An argon or diode laser or a xenon arc is used to coagulate all the blood supply to the tumor.
- Cryotherapy induces damage to the vascular endothelium with secondary thrombosis and infarction of the tumor tissue by rapidly freezing it. It may be used as primary therapy for small peripheral tumors or for small recurrent tumors previously treated with other methods.
- Systemic chemotherapy has become forefront of treatment in the past decade, in the search for globe-preserving measures and to avoid the adverse effects of EBRT therapy. The common indications for chemotherapy for intraocular retinoblastoma include tumors that are large and that cannot be treated with local therapies alone in children with bilateral tumors. It is also used in patients with unilateral disease when the tumors are small, but cannot be controlled with local therapies alone.
- Intra-arterial chemotherapy – Chemotherapeutic drugs are administered locally by a thin catheter threaded through the groin, through the aorta, and the neck, directly into the optic vessels.[41]
- Nanoparticulate chemotherapy – To reduce the adverse effects of systemic therapy, subconjuctival (local) injection of nanoparticle carriers containing chemotherapeutic agents (carboplatin) has been developed, which has shown promising results in the treatment of retinoblastoma in animal models without adverse effects.[42][43]
- Chemoreduction is a combined approach using chemotherapy to initially reduce the size of the tumor, and adjuvant focal treatments, such as transpupillary thermotherapy, to control the tumor.[44][45]
Prognosis[edit]
In the developed world, retinoblastoma has one of the best cure rates of all childhood cancers (95-98%), with more than 90% of sufferers surviving into adulthood. In the UK, around 40 to 50 new cases are diagnosed each year.[46] Good prognosis depends upon early presentation of the child in health facility.[47] Late presentation is associated with a poor prognosis.[48] Survivors of hereditary retinoblastoma have a higher risk of developing other cancers later in life.
Epidemiology[edit]
Retinoblastoma presents with cumulative lifetime incidence rate of one case of retinoblastoma per 18000 to 30000 live births worldwide.[49] A higher incidence is noted in developing countries, which has been attributed to lower socioeconomic status and the presence of human papilloma virus sequences in the retinoblastoma tissue.[50]
Almost 80% of children with retinoblastoma are diagnosed before three years of age and diagnosis in children above six years of age is extremely rare.[51] In the UK, bilateral cases usually present within 14 to 16 months, while diagnosis of unilateral cases peaks between 24 and 30 months.
See also[edit]
References[edit]
- ^ a b c d "Retinoblastoma". St. Jude Children's Research Hospital. Retrieved March 9, 2022.
- ^ a b c Abramson, David H.; Chantada, Guillermo L.; Cobrinik, David; Corson, Timothy W.; Dimaras, Helen; Gallie, Brenda L.; Munier, Francis L.; Njuguna, Festus; Shields, Carol L.; White, Abby; Zhao, Junyang (2015). "Retinoblastoma". Nature Reviews Disease Primers. 1: 15021. doi:10.1038/nrdp.2015.21. PMC 5744255. PMID 27189421.
- ^ a b "Retinoblastoma - Symptoms and causes". Mayo Clinic. Retrieved 2021-03-17.
- ^ a b American Cancer Society (2003). "Chapter 85. Neoplasms of the Eye". Cancer Medicine. Hamilton, Ontario: BC Decker Inc. ISBN 978-1-55009-213-4.
- ^ Naeem, Zishan; Reddy, M. Ashwin; Roelofs, Kelsey A.; Sagoo, Mandeep S.; Warda, Omar (2022). "Retinoblastoma and vision". Eye. 37 (5): 797–808. doi:10.1038/s41433-021-01845-y. PMC 10050411. PMID 34987197. S2CID 245672434.
- ^ Ryan SJ, Schachat AP, Wilkinson CP, Hinton DR, Sadda SR, Wiedemann P (2012-11-01). Retina. Elsevier Health Sciences. p. 2105. ISBN 978-1455737802.
- ^ Dimaras H, Kimani K, Dimba EA, Gronsdahl P, White A, Chan HS, Gallie BL (April 2012). "Retinoblastoma". Lancet. 379 (9824): 1436–1446. doi:10.1016/s0140-6736(11)61137-9. PMID 22414599. S2CID 235331617.
- ^ Elkington AR, Khaw PT (September 1988). "ABC of eyes. Squint". BMJ. 297 (6648): 608–611. doi:10.1136/bmj.297.6648.608. PMC 1834556. PMID 3139234.
- ^ "Retinoblastoma | National Eye Institute". www.nei.nih.gov. Archived from the original on 2022-08-26. Retrieved 2022-08-26.
- ^ "Seen a white glow in a photograph?". chect.org.uk. The Childhood Eye Cancer Trust. 18 January 2017. Retrieved 2022-12-31.
- ^ Balmer, Aubin; Munier, Francis (2007). "Differential diagnosis of leukocoria and strabismus, first presenting signs of retinoblastoma". Clinical Ophthalmology. 1 (4): 431–439. PMC 2704541. PMID 19668520.
- ^ Introduction to White Eye Archived 2011-04-26 at the Wayback Machine, Daisy's Eye Cancer Fund.
- ^ a b c d Du W, Pogoriler J (August 2006). "Retinoblastoma family genes". Oncogene. 25 (38): 5190–5200. doi:10.1038/sj.onc.1209651. PMC 1899835. PMID 16936737.
- ^ Lohmann, Dietmar R.; Gallie, Brenda L. (1993), Adam, Margaret P.; Mirzaa, Ghayda M.; Pagon, Roberta A.; Wallace, Stephanie E. (eds.), "Retinoblastoma", GeneReviews®, Seattle (WA): University of Washington, Seattle, PMID 20301625, retrieved 2022-06-27
- ^ Kivelä T (June 1999). "Trilateral retinoblastoma: a meta-analysis of hereditary retinoblastoma associated with primary ectopic intracranial retinoblastoma". Journal of Clinical Oncology. 17 (6): 1829–1837. doi:10.1200/JCO.1999.17.6.1829. PMID 10561222.
- ^ de Jong MC, Kors WA, de Graaf P, Castelijns JA, Kivelä T, Moll AC (September 2014). "Trilateral retinoblastoma: a systematic review and meta-analysis". The Lancet. Oncology. 15 (10): 1157–1167. doi:10.1016/s1470-2045(14)70336-5. PMID 25126964.
- ^ Harbour J.W., Dean D.C. Rb function in cell-cycle regulation and apoptosis" Nature Cell Biology. 2000;94:E65–E67.
- ^ Parsam VL, Kannabiran C, Honavar S, Vemuganti GK, Ali MJ (December 2009). "A comprehensive, sensitive and economical approach for the detection of mutations in the RB1 gene in retinoblastoma". Journal of Genetics. 88 (4): 517–527. doi:10.1007/s12041-009-0069-z. PMID 20090211. S2CID 10723496.
- ^ Lohmann DR, Gallie BL (2010). "Retinoblastoma". GeneReviews. Seattle, WA: University of Washington. PMID 20301625.
- ^ Ali MJ, Parsam VL, Honavar SG, Kannabiran C, Vemuganti GK, Reddy VA (October 2010). "RB1 gene mutations in retinoblastoma and its clinical correlation". Saudi Journal of Ophthalmology. 24 (4): 119–123. doi:10.1016/j.sjopt.2010.05.003. PMC 3729507. PMID 23960888.
- ^ Rushlow DE, Mol BM, Kennett JY, Yee S, Pajovic S, Thériault BL, et al. (April 2013). "Characterisation of retinoblastomas without RB1 mutations: genomic, gene expression, and clinical studies". The Lancet. Oncology. 14 (4): 327–334. doi:10.1016/S1470-2045(13)70045-7. PMID 23498719.
- ^ Woo CW, Tan F, Cassano H, Lee J, Lee KC, Thiele CJ (February 2008). "Use of RNA interference to elucidate the effect of MYCN on cell cycle in neuroblastoma". Pediatric Blood & Cancer. 50 (2): 208–212. doi:10.1002/pbc.21195. PMID 17420990. S2CID 22765085.
- ^ Stenfelt S, Blixt MK, All-Ericsson C, Hallböök F, Boije H (November 2017). "Heterogeneity in retinoblastoma: a tale of molecules and models". Clinical and Translational Medicine. 6 (1): 42. doi:10.1186/s40169-017-0173-2. PMC 5680409. PMID 29124525.
- ^ Lewis R (March 19, 2013). "Some Aggressive Retinoblastomas Lack RB1 Mutations". Medscape Online. Archived from the original on September 19, 2017.
- ^ Bowman, Richard; Evans, Jennifer R.; Gilbert, Clare; Gordon, Iris; Gupta, Sunchita; Malik, Aeesha NJ; Mariotti, Silvio (2022). "Universal newborn eye screening: a systematic review of the literature and review of international guidelines" (PDF). Journal of Global Health. 12: 12003. doi:10.7189/jogh.12.12003. PMC 9586142. PMID 36269293. Retrieved 2022-12-31.
- ^ Abramson, David H.; Barnea, Dana; Bosscha, Machted I.; Chantada, Guillermo; de Graaf, Pim; Dommering, Charlotte J.; Dunkel, Ira J.; Fabius, Armida W.M.; Francis, Jasmine H.; Greer, Mary-Louise G.; Kleinerman, Ruth A.; Kors, Wijnanda A.; Laughlin, Suzanne; Moll, Annette C.; Morton, Lindsay M.; Novetsky Friedman, Danielle; Oeffinger, Kevin C.; Temming, Petra; Tonorezos, Emily S.; Tucker, Margaret A.; van Leeuwen, Flora E.; Walsh, Michael F. (2020). "Recommendations for long-term follow-up of adults with heritable retinoblastoma". Ophthalmology. 127 (11): 1549–1557. doi:10.1016/j.ophtha.2020.05.024. PMC 7606265. PMID 32422154.
- ^ MacCarthy A, Birch JM, Draper GJ, Hungerford JL, Kingston JE, Kroll ME, et al. (January 2009). "Retinoblastoma in Great Britain 1963-2002". The British Journal of Ophthalmology. 93 (1): 33–37. doi:10.1136/bjo.2008.139618. PMID 18838413. S2CID 27049728.
- ^ de Jong MC, de Graaf P, Noij DP, Göricke S, Maeder P, Galluzzi P, et al. (May 2014). "Diagnostic performance of magnetic resonance imaging and computed tomography for advanced retinoblastoma: a systematic review and meta-analysis". Ophthalmology. 121 (5): 1109–1118. doi:10.1016/j.ophtha.2013.11.021. PMID 24589388.
- ^ Sehu WK, Lee W (2005). Ophthalmic pathology : an illustrated guide for clinicians. Malden: Blackwell Publishing. p. 262. ISBN 978-0-7279-1779-9.
- ^ Kumar V, Abbas AK, Fausto N (1999). Robbins pathologic basis of disease (6th ed.). Philadelphia: Saunders. p. 1442. ISBN 978-0-7216-0187-8.
- ^ Schüler A, Weber S, Neuhäuser M, Jurklies C, Lehnert T, Heimann H, et al. (March 2005). "Age at diagnosis of isolated unilateral retinoblastoma does not distinguish patients with and without a constitutional RB1 gene mutation but is influenced by a parent-of-origin effect". European Journal of Cancer. 41 (5): 735–740. doi:10.1016/j.ejca.2004.12.022. PMID 15763650.
- ^ a b Rushlow D, Piovesan B, Zhang K, Prigoda-Lee NL, Marchong MN, Clark RD, Gallie BL (May 2009). "Detection of mosaic RB1 mutations in families with retinoblastoma". Human Mutation. 30 (5): 842–851. doi:10.1002/humu.20940. PMID 19280657. S2CID 31887184.
- ^ a b Richter S, Vandezande K, Chen N, Zhang K, Sutherland J, Anderson J, et al. (February 2003). "Sensitive and efficient detection of RB1 gene mutations enhances care for families with retinoblastoma". American Journal of Human Genetics. 72 (2): 253–269. doi:10.1086/345651. PMC 379221. PMID 12541220.
- ^ Canadian Ophthalmological Society (December 2009). "National Retinoblastoma Strategy Canadian Guidelines for Care" (PDF). Canadian Journal of Ophthalmology. 44 (suppl.2): S1-88. doi:10.3129/i09-194. PMID 20237571. Archived from the original (PDF) on 2011-09-29.
- ^ Dimaras H, Corson TW (January 2019). "Retinoblastoma, the visible CNS tumor: A review". Journal of Neuroscience Research. 97 (1): 29–44. doi:10.1002/jnr.24213. PMC 6034991. PMID 29314142.
- ^ a b c Chawla B, Jain A, Azad R (September 2013). "Conservative treatment modalities in retinoblastoma". Indian Journal of Ophthalmology. 61 (9): 479–485. doi:10.4103/0301-4738.119424. PMC 3831762. PMID 24104705.
- ^ a b Fabian ID, Reddy A, Sagoo MS (2018). "Classification and staging of retinoblastoma". Community Eye Health. 31 (101): 11–13. PMC 5998397. PMID 29915461.
- ^ a b Chintagumpala M, Chevez-Barrios P, Paysse EA, Plon SE, Hurwitz R (October 2007). "Retinoblastoma: review of current management". The Oncologist. 12 (10): 1237–1246. CiteSeerX 10.1.1.585.5448. doi:10.1634/theoncologist.12-10-1237. PMID 17962617. S2CID 15465073.
- ^ Li WL, Buckley J, Sanchez-Lara PA, Maglinte DT, Viduetsky L, Tatarinova TV, et al. (July 2016). "A Rapid and Sensitive Next-Generation Sequencing Method to Detect RB1 Mutations Improves Care for Retinoblastoma Patients and Their Families". The Journal of Molecular Diagnostics. 18 (4): 480–493. doi:10.1016/j.jmoldx.2016.02.006. PMC 5820122. PMID 27155049.
- ^ Roarty JD, McLean IW, Zimmerman LE (November 1988). "Incidence of second neoplasms in patients with bilateral retinoblastoma". Ophthalmology. 95 (11): 1583–1587. doi:10.1016/s0161-6420(88)32971-4. PMID 3211467.
- ^ Shields CL, Ramasubramanian A, Rosenwasser R, Shields JA (September 2009). "Superselective catheterization of the ophthalmic artery for intraarterial chemotherapy for retinoblastoma". Retina. 29 (8): 1207–1209. doi:10.1097/IAE.0b013e3181b4ce39. PMID 19734768. S2CID 47557934.
- ^ Shome D, Poddar N, Sharma V, Sheorey U, Maru GB, Ingle A, et al. (December 2009). "Does a nanomolecule of Carboplatin injected periocularly help in attaining higher intravitreal concentrations?". Investigative Ophthalmology & Visual Science. 50 (12): 5896–5900. doi:10.1167/iovs.09-3914. PMID 19628744. S2CID 7361361.
- ^ Kang SJ, Durairaj C, Kompella UB, O'Brien JM, Grossniklaus HE (August 2009). "Subconjunctival nanoparticle carboplatin in the treatment of murine retinoblastoma". Archives of Ophthalmology. 127 (8): 1043–1047. doi:10.1001/archophthalmol.2009.185. PMC 2726977. PMID 19667343.
- ^ Fabian ID, Johnson KP, Stacey AW, Sagoo MS, Reddy MA (June 2017). "Focal laser treatment in addition to chemotherapy for retinoblastoma". The Cochrane Database of Systematic Reviews. 2017 (6): CD012366. doi:10.1002/14651858.CD012366.pub2. PMC 6481366. PMID 28589646.
- ^ Shields CL, Mashayekhi A, Cater J, Shelil A, Ness S, Meadows AT, Shields JA (June 2005). "Macular retinoblastoma managed with chemoreduction: analysis of tumor control with or without adjuvant thermotherapy in 68 tumors". Archives of Ophthalmology. 123 (6): 765–773. doi:10.1001/archopht.123.6.765. PMID 15955977.
- ^ Beddard, N; McGeechan, GJ; Taylor, J; Swainston, K (2019). "Childhood eye cancer from a parental perspective: The lived experience of parents with children who have had retinoblastoma". European Journal of Cancer Care. 29 (2): e13209. doi:10.1111/ecc.13209. PMID 31845431. S2CID 196549559.
- ^ Concise ophthalmology : for medical students (4th ed.). Karachi, Pakistan: Paramount Books. 2014. pp. 80–81. ISBN 9789696370017.
- ^ Rai P, Shah IA, Narsani AK, Lohana MK, Memon MK, Memon MA (2009). "Too late presentation of 53 patients with retinoblastoma". International Journal of Ophthalmology. 9 (2): 227–230.
- ^ Abramson DH, Schefler AC (December 2004). "Update on retinoblastoma". Retina. 24 (6): 828–848. doi:10.1097/00006982-200412000-00002. PMID 15579980. S2CID 26883037.
- ^ Orjuela M, Castaneda VP, Ridaura C, Lecona E, Leal C, Abramson DH, et al. (October 2000). "Presence of human papilloma virus in tumor tissue from children with retinoblastoma: an alternative mechanism for tumor development". Clinical Cancer Research. 6 (10): 4010–4016. PMID 11051250.
- ^ Abramson DH, Frank CM, Susman M, Whalen MP, Dunkel IJ, Boyd NW (March 1998). "Presenting signs of retinoblastoma". The Journal of Pediatrics. 132 (3 Pt 1): 505–508. doi:10.1016/s0022-3476(98)70028-9. PMID 9544909.