
Spongy degeneration of the central nervous system
| Spongy Degeneration of the Central Nervous System | |
|---|---|
| Other names | Canavan's disease, Van Bogaert-Bertrand type, Aspartoacylase deficiency |
 | |
| Magnetic resonance imaging scans showing dysmyelination, a possible indicator of Canavan's disease | |
| Specialty | Neurology |
| Symptoms | Hypotonia, macrocephaly, loss of vision, motor reflex defects, difficulties in breathing and swallowing |
| Usual onset | 3-6 months of age |
| Duration | Terminal |
| Types | Infantile, congenital, juvenile |
| Causes | Genetic (Autosomal recessive) |
| Risk factors | Family history (genetics) |
| Diagnostic method | Neuroimaging, urine examination |
| Prevention | DNA analysis, prenatal analysis |
| Treatment | Palliative measures only |
| Medication | N/A |
Spongy degeneration of the central nervous system, also known as Canavan's disease, Van Bogaert-Bertrand type or Aspartoacylase (AspA) deficiency, is a rare autosomal recessive neurodegenerative disorder.[1] It belongs to a group of genetic disorders known as leukodystrophies,[1] where the growth and maintenance of myelin sheath in the central nervous system (CNS) are impaired.[2] There are three types of spongy degeneration: infantile, congenital and juvenile, with juvenile being the most severe type.[3] Common symptoms in infants include lack of motor skills, weak muscle tone, and macrocephaly.[4] It may also be accompanied by difficulties in feeding and swallowing, seizures and sleep disturbances.[4] Affected children typically die before the age of 10, but life expectancy can vary.[5]
The cause of spongy degeneration of the CNS is the mutation in a gene coding for aspartoacylase (AspA), an enzyme that hydrolyzes N-acetyl aspartic acid (NAA).[6] In the absence of AspA, NAA accumulates and results in spongy degeneration.[7] The exact pathophysiological causes of the disease are currently unclear, but there are developing theories.[8] Spongy degeneration can be diagnosed with neuroimaging techniques and urine examination.[9] There is no current treatment for spongy degeneration, but research utilising gene therapy to treat the disease is underway.[8] Spongy degeneration is found to be more prevalent among Ashkenazi Jews, with an incidence of 1/6000 amongst this ethnic group.[10]
Clinical Symptoms[edit]
Spongy Degeneration of the CNS is classified into three types: infantile, juvenile and congenital; based on the age of onset and severity of symptoms.
Infantile Type[edit]
The infantile type is the most common type of spongy degeneration of the CNS.[11] Usually, affected infants appear normal for the first few months of life.[12] The age of onset is around 6 months, where infants begin to develop noticeable psychomotor defects.[12] Various motor skills such as turning over and stabilising head movements are affected.[11] Hypotonia and macrocephaly are also observed in the first few months.[13]
During the latter part of the first year, most children's eyes fail to respond to visual stimuli, with episodic saccadic eye movements observed, rendering most children blind in the second year.[5]
The symptoms in the terminal stage of disease development are sweating, emesis, hyperthermia, seizures, and hypotension, which usually results in the death of the child.[13] Life expectancies of affected infants vary, but most infants do not live past the age of ten.[5]

Congenital Type[edit]
The age of onset is typically a few days after birth in the congenital type. Pregnancy and delivery are not affected and the child is born with a normal appearance and no health issues.[12] However, affected infants may become lethargic in the following days and find movements such as sucking and swallowing difficult.[14] As the disease progresses, patients may have decreased muscle tone and inactivation of Moro reflex, also known as startle reflex.[12] This may lead to the development of Cheyne Stokes respiration after a few weeks or even days after delivery, which may be fatal.[12]
Juvenile Type[edit]
The age of onset of the juvenile type is around five years of age. Most patients with the juvenile type survive until late adolescence.[15] Affected toddlers typically develop progressive cerebellar syndrome and mental deterioration, which is followed by vision loss, optic atrophy, and generalised spasticity.[16] Unlike the infantile form, there is no macrocephaly exhibited.[12]
Pathophysiology[edit]
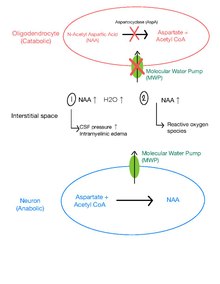
Although the pathophysiological causes of CD symptoms are still unclear, there are developing theories on the causes of myelination issues, gelatinous cortical white matter and seizures.[8]
Issues in Myelination[edit]
Molecular Water Pump (MWP) and Osmolyte Imbalance[edit]
Increased cerebrospinal fluid (CSF) pressure and intramyelinic edema in CD patients suggest the existence of an efficient MWP in the brain.[17][8] The MWP is a membrane protein responsible for pumping water molecules, along with dissolved NAA molecules, from the intraneuronal space to the interstitial space.[10] In healthy individuals, NAA is first transported down the concentration gradient through the MWP from neurons to the interstitial space and subsequently hydrolyzed by AspA in neighbouring oligodendrocytes.[10]
In patients with CD, it is theorized that AspA deficiency causes accumulation of NAA in the interstitial space, inducing an osmolyte imbalance and accumulation of water in the interstitial space.[8] This increases hydrostatic pressure between interlamellar spaces and extracellular periaxonal and parenchymatous space, loosening the tight junctions between them, thus causing intramyelinic edema.[18] Subsequent demyelination possibly contributes to vacant spaces in the white matter or spongy degeneration.[8]
Dysmyelination[edit]
NAA-derived acetates are involved in the synthesis of fatty acids, which are subsequently incorporated into myelin lipids.[3][8] It is hypothesized that in CD patients, AspA deficiency reduces NAA-derived acetates, and consequently decreases the synthesis of myelin-associated lipids.[6] This leads to dysmyelination, which promotes the formation vacuoles in interstitial space and spongy degeneration.[8] However, it has been shown that spongy degeneration is not directly caused by the disrupted synthesis of myelin.[19] Animal models show that myelination may still occur in AspA lacking species, possibly due to parallel pathways for myelination during the initial stages of myelinogenesis.[19]
Protein Folding and Stabilization[edit]
Deficiency of AspA lowers acetyl coenzyme A (CoA) expression in cells, which may be responsible for stabilization and correct folding of proteins.[20] This leads to protein degradation, with a particularly large effect in oligodendrocytes.[20] In animal studies of AspA deficient species, protein degradation in oligodendrocytes has been shown to cause severe loss in myelin proteins.[21]
Gelatinous Subcortical White Matter[edit]
The deficiency in AspA, which is vital in oligodendrocytes to produce NAA derived acetate, leads to a lack of regulation in the genetic structure and expression in these cells.[1] This results in the death of oligodendrocytes, hence induces neuronal injury and the formation of vacuoles in the subcortical matter.[22] These vacuoles contribute to the formation of gelatinous-textured subcortical white matter found in many CD patients.[22]
Seizures and Neurodegeneration[edit]
The pathophysiological causes of seizures and neurodegeneration in CD patients are likely due to oxidative stress generated by NAA accumulation.[23] It is postulated that NAA promotes oxidative stress through promoting reactive oxygen species, as well as reducing non-enzymatic antioxidant defenses.[23] NAA also affects multiple antioxidant enzymes, such as catalase and glutathione peroxidase, impairing the detoxification of hydrogen peroxide.[24] Recent animal studies have shown the chronic oxidative stress may cause dysfunction in mitochondria, rendering the brain more susceptible to epileptic seizures.[24][25]
Diagnosis[edit]
Canavan's disease is initially recognized by the appearance of symptoms, yet further examinations are needed for definitive diagnosis. Neuroimaging techniques such as Computed Tomography (CT) scan or Magnetic Resonance imaging (MRI) are typically used to detect the presence of degenerative subcortical white matter.[26] Microscopy of the cerebrospinal fluid can also be used for diagnosis, where swollen astrocytes with distorted and elongated mitochondria can be seen in patients.[5]
Urine examinations are used to differentiate CD patients from other neurodegenerative disorders with similar morphology, such as Alexander diseases and Tay-Sachs diseases (which similarly exhibit macrocephaly), as patients with CD uniquely display increased excretion of NAA.[5][13]
Prevention[edit]

DNA analysis is generally used to determine if parents are carriers of the mutant gene.[27] Prenatal diagnosis through either DNA analysis or determination of NAA in amniotic fluid (which would be increased in an affected pregnancy) can also be used when DNA analysis cannot be performed on parents.[28] It has been observed that there is an abnormally high carrier rate in the Ashkenazi Jewish population.[4] The risk of their offspring having spongy degeneration is one in four if both parents are carriers of the mutant gene.[28]
Treatment methods[edit]
There are currently no specific forms of treatment known for spongy degeneration of the CNS.[29] Certain treatment modules are under experimental trials and current patients are supported by palliative measures, all of which are described below.
Current palliative measures[edit]
Current patients are supported by the care guidelines for other paediatric neurodegenerative diseases.[30] For patients with respiratory issues, suction machines are used to clear mucous from the upper airway of the lungs.[8] Oxygen concentrators are also administered for airway clearance and continuous supply of air to aid breathing.[8] As for infants with hypotonia, it is addressed by the provision of positioning equipment like specialized strollers, bath chairs and feeder seats.[31]
Possible treatment modules under development[edit]
Intraperitoneal injections of lipoic acid[edit]
Lipoic acid (which can cross the blood brain barrier), has recently been trialed in preclinical studies, where it has been injected into tremor rats intraperitoneally.[32] Tremor rats are deemed as the naturally occurring model for spongy degeneration of the CNS as NAA induces oxidative stress.[33] Positive results have emerged from these studies, suggesting that lipoic acid may be a possible approach for symptomatic treatments.[32]
Intraperitoneal lithium administration[edit]
A possible treatment is to employ neuroprotective techniques to offset the neurological damage in the CNS caused by the accumulation of NAA.[8] One potential treatment that has been identified is lithium, which has been observed to induce neuroprotective effects in dementia patients.[34] Administration of intraperitoneal lithium has been tested in both tremor and wild-type rats, causing a decrease in NAA levels in both species.[35] In human trials, NAA levels in patient's brain and urine was found to drop after one year of treatment.[29] This is coupled with the elevation of alertness and visual tracking.[29] However. CD symptoms including axial hypotonia and spastic diplegia remained.[8]

Gene therapy[edit]
Since CD arises from a monogenic defect and is localized in the CNS, gene replacement therapy is a potential treatment.[8] This therapy involves replacing the mutant gene of the disease with a fully functional gene using a vector, which transports therapeutic DNA into cells, allowing cells to produce AspA.[36] Adeno-associated Viruses (AAVs) are widely used as vectors for gene therapy.[8] They are adopted as they do not replicate themselves and are almost non-toxic.[8] There are two serotypes used for the treatment: AAV2 and AAV9.[37] The difference of the stereotypes is that AAV2 is limited by blood-brain-barrier (BBB), whilst AAV9 can cross the BBB, allowing for treatment even at the later stages of the disease.[38] However, current research shows that AAVs may trigger unwanted immune responses in infants and have limited gene encapsulating capacity.[39]
Epidemiology[edit]
Spongy degeneration of the CNS is pan-ethnic, due to its prevalence among Ashkenazi Jews. There are two common mutations found among them: missense mutation (Glu285AIa) and nonsense mutation (Tyr231X).[40] In the missense mutation, there is a substitution of glutamic acid to alanine.[41] As for the nonsense mutation, the tyrosine codon is replaced by a termination codon.[41] Genetic screening reveals that around 1 in 40 healthy Jews are carriers and the incidence of this disease in this population is as high as 1 in 6000.[10]
History[edit]
The first case of spongy degeneration of the CNS was reported in 1928 by Globus and Strauss,[42] who designated the case as Schilder's disease, a term for diffuse myelinoclastic sclerosis.[43][44][45] In 1931, Canavan reported a case where the megalencephaly of brain degeneration is different from that caused by a tumour.[46] However, she failed to recognize the spongy alterations that suggest a unique pathological cause that distinguishes her case from Schilder's disease.[47] Later in 1937, Eislebergl reported six cases from Jewish families and discovered the familial characteristics of spongy degeneration, but she classified these cases as Krabbe's sclerosis.[47][48] It was not until 1949 when Van Bogaert and Bertrand reported five cases from Jewish families, whereupon further pathological analysis confirmed that spongy degeneration is the nosologic entity.[47]
References[edit]
- ^ a b c Baslow MH, Guilfoyle DN (April 2013). "Canavan disease, a rare early-onset human spongiform leukodystrophy: insights into its genesis and possible clinical interventions". Biochimie. 95 (4): 946–56. doi:10.1016/j.biochi.2012.10.023. PMID 23151389.
- ^ "Canavan Disease". NORD (National Organization for Rare Disorders). Retrieved 2021-03-31.
- ^ a b Namboodiri AM, Peethambaran A, Mathew R, Sambhu PA, Hershfield J, Moffett JR, Madhavarao CN (June 2006). "Canavan disease and the role of N-acetylaspartate in myelin synthesis". Molecular and Cellular Endocrinology. 252 (1–2): 216–23. doi:10.1016/j.mce.2006.03.016. PMID 16647192. S2CID 12255670.
- ^ a b c Feigenbaum A, Moore R, Clarke J, Hewson S, Chitayat D, Ray PN, Stockley TL (January 2004). "Canavan disease: carrier-frequency determination in the Ashkenazi Jewish population and development of a novel molecular diagnostic assay". American Journal of Medical Genetics. Part A. 124A (2): 142–7. doi:10.1002/ajmg.a.20334. PMID 14699612. S2CID 25981659.
- ^ a b c d e Matalon RM, Michals-Matalon K (March 2000). "Spongy degeneration of the brain, Canavan disease: biochemical and molecular findings". Frontiers in Bioscience. 5: D307-11. doi:10.2741/matalon. PMID 10704428.
- ^ a b Madhavarao CN, Arun P, Moffett JR, Szucs S, Surendran S, Matalon R, et al. (April 2005). "Defective N-acetylaspartate catabolism reduces brain acetate levels and myelin lipid synthesis in Canavan's disease". Proceedings of the National Academy of Sciences of the United States of America. 102 (14): 5221–6. Bibcode:2005PNAS..102.5221M. doi:10.1073/pnas.0409184102. PMC 555036. PMID 15784740.
- ^ Surendran S, Michals-Matalon K, Quast MJ, Tyring SK, Wei J, Ezell EL, Matalon R (March 2006). "Canavan disease: a monogenic trait with complex genomic interaction". Molecular Genetics and Metabolism. 80 (1–2): 74–80. doi:10.1016/j.ymgme.2003.08.015. PMID 14567959.
- ^ a b c d e f g h i j k l m n o Ahmed SS, Gao G (2014). Zschocke J, Baumgartner M, Morava E, Patterson M (eds.). "Making the White Matter Matters: Progress in Understanding Canavan's Disease and Therapeutic Interventions Through Eight Decades". JIMD Reports. 19. Berlin, Heidelberg: Springer Berlin Heidelberg: 11–22. doi:10.1007/8904_2014_356. ISBN 978-3-662-46189-1. PMC 4501231. PMID 25604619.
- ^ Matalon R, Michals K, Kaul R (October 1995). "Canavan disease: from spongy degeneration to molecular analysis". The Journal of Pediatrics. 127 (4): 511–7. doi:10.1016/S0022-3476(95)70105-2. PMID 7562269.
- ^ a b c d Bokhari MR, Samanta D, Bokhari SR (2021). Canavan Disease. Treasure Island (FL): StatPearls Publishing. PMID 28613566.
{{cite book}}:|work=ignored (help) - ^ a b Matalon R, Delgado L, Michals-Matalon K (January 2020). "Chapter 66 - Canavan disease". In Rosenberg RN, Pascual JM (eds.). Rosenberg's Molecular and Genetic Basis of Neurological and Psychiatric Disease (Sixth ed.). Academic Press. pp. 909–916. ISBN 978-0-12-813955-4.
- ^ a b c d e f Adachi M, Schneck L, Cara J, Volk BW (September 1973). "Spongy degeneration of the central nervous system (van Bogaert and Bertrand type; Canavan's disease). A review". Human Pathology. 4 (3): 331–47. doi:10.1016/s0046-8177(73)80098-x. PMID 4593851.
- ^ a b c Matalon R, Delgado L, Michals-Matalon K (1993). Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJ, Mirzaa G, Amemiya A (eds.). Canavan Disease. Seattle (WA): University of Washington, Seattle. PMID 20301412.
{{cite book}}:|work=ignored (help) - ^ Kantor B, McCown T, Leone P, Gray SJ (2014-01-01). Clinical applications involving CNS gene transfer. Advances in Genetics. Vol. 87. pp. 71–124. doi:10.1016/B978-0-12-800149-3.00002-0. ISBN 9780128001493. PMC 4518844. PMID 25311921.
- ^ Traeger EC, Rapin I (March 1998). "The clinical course of Canavan disease". Pediatric Neurology. 18 (3): 207–12. doi:10.1016/s0887-8994(97)00185-9. PMID 9568915.
- ^ "Canavan Disease - MeSH - NCBI". www.ncbi.nlm.nih.gov. Retrieved 2021-04-13.
- ^ Zeuthen T (2000). "Molecular water pumps". Reviews of Physiology, Biochemistry and Pharmacology. 141. Berlin, Heidelberg: Springer Berlin Heidelberg: 97–151. doi:10.1007/bfb0119578. ISBN 978-3-540-66627-1. PMID 10916424.
- ^ "Brain - Intramyelinic Edema - Nonneoplastic Lesion Atlas". ntp.niehs.nih.gov. Retrieved 2021-04-13.
- ^ a b Wang J, Leone P, Wu G, Francis JS, Li H, Jain MR, et al. (January 2009). "Myelin lipid abnormalities in the aspartoacylase-deficient tremor rat". Neurochemical Research. 34 (1): 138–48. doi:10.1007/s11064-008-9726-5. PMC 4640699. PMID 18478328.
- ^ a b Spange S, Wagner T, Heinzel T, Krämer OH (January 2009). "Acetylation of non-histone proteins modulates cellular signalling at multiple levels". The International Journal of Biochemistry & Cell Biology. 41 (1): 185–98. doi:10.1016/j.biocel.2008.08.027. PMID 18804549.
- ^ Kumar S, Biancotti JC, Matalon R, de Vellis J (November 2009). "Lack of aspartoacylase activity disrupts survival and differentiation of neural progenitors and oligodendrocytes in a mouse model of Canavan disease". Journal of Neuroscience Research. 87 (15): 3415–27. doi:10.1002/jnr.22233. PMID 19739253. S2CID 44554774.
- ^ a b "Aspartoacylase Deficiency (Canavan Disease) | The Online Metabolic and Molecular Bases of Inherited Disease | OMMBID | McGraw-Hill Medical". ommbid.mhmedical.com. Retrieved 2021-04-15.
- ^ a b Francis JS, Strande L, Markov V, Leone P (September 2012). "Aspartoacylase supports oxidative energy metabolism during myelination". Journal of Cerebral Blood Flow and Metabolism. 32 (9): 1725–36. doi:10.1038/jcbfm.2012.66. PMC 3434629. PMID 22617649.
- ^ a b Pederzolli CD, Mescka CP, Scapin F, Rockenbach FJ, Sgaravatti AM, Sgarbi MB, et al. (August 2007). "N-acetylaspartic acid promotes oxidative stress in cerebral cortex of rats". International Journal of Developmental Neuroscience. 25 (5): 317–24. doi:10.1016/j.ijdevneu.2007.04.002. PMID 17604935. S2CID 41719397.
- ^ Chuang YC (March 2010). "Mitochondrial dysfunction and oxidative stress in seizure-induced neuronal cell death". Acta Neurologica Taiwanica. 19 (1): 3–15. PMID 20711885.
- ^ Sreenivasan P, Purushothaman KK (January 2013). "Radiological clue to diagnosis of Canavan disease". Indian Journal of Pediatrics. 80 (1): 75–7. doi:10.1007/s12098-012-0794-9. PMID 22660905. S2CID 35979708.
- ^ "Canavan Disease, DNA Analysis - Tests - GTR - NCBI". www.ncbi.nlm.nih.gov. Retrieved 2021-03-31.
- ^ a b "Canavan Disease Information Page | National Institute of Neurological Disorders and Stroke". www.ninds.nih.gov. Retrieved 2021-03-31.
- ^ a b c Hoshino H, Kubota M (August 2014). "Canavan disease: clinical features and recent advances in research". Pediatrics International. 56 (4): 477–83. doi:10.1111/ped.12422. PMID 24977939. S2CID 206261733.
- ^ Hunt A, Burne R (January 1995). "Medical and nursing problems of children with neurodegenerative disease". Palliative Medicine. 9 (1): 19–26. doi:10.1177/026921639500900104. PMID 7719515. S2CID 20328927.
- ^ Bakewell J (August 2007). "Choosing support equipment in children's therapy". International Journal of Therapy and Rehabilitation. 14 (8): 379–381. doi:10.12968/ijtr.2007.14.8.24358. ISSN 1741-1645.
- ^ a b Roscoe RB, Elliott C, Zarros A, Baillie GS (July 2016). "Non-genetic therapeutic approaches to Canavan disease" (PDF). Journal of the Neurological Sciences. 366: 116–124. doi:10.1016/j.jns.2016.05.012. PMID 27288788. S2CID 329155.
- ^ Surendran S, Bhatnagar M (June 2011). "Upregulation of N-acetylaspartic acid induces oxidative stress to contribute in disease pathophysiology". The International Journal of Neuroscience. 121 (6): 305–9. doi:10.3109/00207454.2011.558225. PMID 21348802. S2CID 5406213.
- ^ Matsunaga S, Kishi T, Annas P, Basun H, Hampel H, Iwata N (2015). "Lithium as a Treatment for Alzheimer's Disease: A Systematic Review and Meta-Analysis". Journal of Alzheimer's Disease. 48 (2): 403–10. doi:10.3233/JAD-150437. PMID 26402004.
- ^ Lienhard U, Sass J (2011-01-01). "Canavan Disease: A Neurometabolic Disease Caused By Aspartoacylase Deficiency". Journal of Pediatric Sciences. 3 (1): 1–11. doi:10.17334/jps.44686 (inactive 31 January 2024).
{{cite journal}}: CS1 maint: DOI inactive as of January 2024 (link) - ^ Naso MF, Tomkowicz B, Perry WL, Strohl WR (August 2017). "Adeno-Associated Virus (AAV) as a Vector for Gene Therapy". BioDrugs. 31 (4): 317–334. doi:10.1007/s40259-017-0234-5. PMC 5548848. PMID 28669112.
- ^ Wang D, Tai PW, Gao G (May 2019). "Adeno-associated virus vector as a platform for gene therapy delivery". Nature Reviews. Drug Discovery. 18 (5): 358–378. doi:10.1038/s41573-019-0012-9. PMC 6927556. PMID 30710128.
- ^ Liu D, Zhu M, Zhang Y, Diao Y (January 2021). "Crossing the blood-brain barrier with AAV vectors". Metabolic Brain Disease. 36 (1): 45–52. doi:10.1007/s11011-020-00630-2. ISSN 0885-7490. PMID 33201426. S2CID 226986888.
- ^ Chen W, Hu Y, Ju D (August 2020). "Gene therapy for neurodegenerative disorders: advances, insights and prospects". Acta Pharmaceutica Sinica B. 10 (8): 1347–1359. doi:10.1016/j.apsb.2020.01.015. PMC 7488363. PMID 32963936.
- ^ Bass NE, Debrosse SD (2014-01-01). "Canavan's Disease". Encyclopedia of the Neurological Sciences. Academic Press. pp. 577–579. doi:10.1016/B978-0-12-385157-4.00088-9. ISBN 9780123851581.
- ^ a b Kaul R, Gao GP, Aloya M, Balamurugan K, Petrosky A, Michals K, Matalon R (July 1994). "Canavan disease: mutations among Jewish and non-Jewish patients". American Journal of Human Genetics. 55 (1): 34–41. PMC 1918221. PMID 8023850.
- ^ Globus JH, Strauss I (1928-12-01). "Progressive Degenerative Subcortical Encephalopathy". Archives of Neurology & Psychiatry. 20 (6): 1190–1228. doi:10.1001/archneurpsyc.1928.02210180041003. ISSN 0096-6754.
- ^ Klugmann, Matthias; Leichtlein, Claudia B. (2006). "Clinical Trials of Gene Therapy for Canavan Disease – I. The Nosology of Canavan Disease". In Kaplitt, Michael G.; During, Matthew J. (eds.). Gene Therapy of the Central Nervous System. pp. 303–316. doi:10.1016/B978-012397632-1/50024-1. ISBN 978-0-12-397632-1.
- ^ Bertorini, Tulio E.; Perez, Angel (2014). "Neurologic complications of disorders of the adrenal glands". In Biller, José; Ferro, José M. (eds.). Handbook of Clinical Neurology: Neurologic Aspects of Systemic Disease Part II. Vol. 120. pp. 749–771. doi:10.1016/B978-0-7020-4087-0.00050-4. ISBN 9780702040870. PMID 24365350.
- ^ Poser, CM; Goutières, F; Carpentier, MA; Aicardi, J (January 1986). "Schilder's myelinoclastic diffuse sclerosis". Pediatrics. 77 (1): 107–112. PMID 3940347. Erratum in: Pediatrics. 78(1): 138, (July 1986).
The term "Schilder's disease" has been used to describe conditions as disparate as adrenoleukodystrophy, myelinoclastic diffuse sclerosis... The eponymic designation should be reserved for instances of myelinoclastic diffuse sclerosis that correspond to the case described by Schilder in 1912.
- ^ Canavan MM (February 1931). "Schilder's encephalitis periaxialis diffusa: Report of a case in a child aged sixteen and one-half months". Archives of Neurology & Psychiatry. 25 (2): 299–308. doi:10.1001/archneurpsyc.1931.02230020085005.
- ^ a b c Adachi M (1967). "Studies on Spongy Degeneration of the Central Nervous System (Van Bogaert–Bertrand Type)". Inborn Disorders of Sphingolipid Metabolism. Elsevier. pp. 129–147. doi:10.1016/b978-1-4831-9855-2.50013-7. ISBN 978-1-4831-9855-2.
- ^ Eiselsberg F (1937-06-01). "Über frühkindliche familiäre diffuse Hirnsklerose". Zeitschrift für Kinderheilkunde (in German). 58 (6): 702–725. doi:10.1007/BF02249721. ISSN 1432-1076. S2CID 31242183.