
Paraganglioma
| Paraganglioma | |
|---|---|
| Other names | Chemodectoma, paraganglioma, carotid body tumour, glomus cell tumour |
 | |
| Micrograph of a carotid body tumor (a type of paraganglioma) | |
| Specialty | Oncology |
A paraganglioma is a rare neuroendocrine neoplasm that may develop at various body sites (including the head, neck, thorax and abdomen). When the same type of tumor is found in the adrenal gland, they are referred to as a pheochromocytoma. They are rare tumors, with an overall estimated incidence of 1 in 300,000.[1] There is no test that determines benign from malignant tumors; long-term follow-up is therefore recommended for all individuals with paraganglioma.[2]
Signs and symptoms
[edit]Most paragangliomas are asymptomatic, present as a painless mass, or create symptoms such as hypertension, tachycardia, headache, and palpitations.[3] While all contain neurosecretory granules, only in 1–3% of cases is secretion of hormones such as catecholamines abundant enough to be clinically significant; in that case manifestations often resemble those of pheochromocytomas (intra-medullary paraganglioma).[citation needed]
Genetics
[edit]About 75% of paragangliomas are sporadic; the remaining 25% are hereditary (and have an increased likelihood of being multiple and of developing at an earlier age). Mutations of the genes for the succinate dehydrogenase, SDHD (previously known as PGL1), SDHA, SDHC (previously PGL3) and SDHB have been identified as causing familial head and neck paragangliomas. Mutations of SDHB play an important role in familial adrenal pheochromocytoma and extra-adrenal paraganglioma (of abdomen and thorax), although there is considerable overlap in the types of tumors associated with SDHB and SDHD gene mutations. Paragangliomas may also occur in MEN type 2A and 2B. Other genes related to familial paraganglioma are SDHAF2,[4] VHL, NF1, TMEM127,[5] MAX[6] and SLC25A11.[7]
Pathology
[edit]

The paragangliomas appear grossly as sharply circumscribed polypoid masses and they have a firm to rubbery consistency. They are highly vascular tumors and may have a deep red color.[citation needed]
On microscopic inspection, the tumor cells are readily recognized. Individual tumor cells are polygonal to oval and are arranged in distinctive cell balls, called Zellballen.[8] These cell balls are separated by fibrovascular stroma and surrounded by sustentacular cells.[citation needed]
By light microscopy, the differential diagnosis includes related neuroendocrine tumors, such as carcinoid tumor, neuroendocrine carcinoma, and medullary carcinoma of the thyroid.[citation needed]
With immunohistochemistry, the chief cells located in the cell balls are positive for chromogranin, synaptophysin, neuron specific enolase, serotonin, neurofilament and Neural cell adhesion molecule; they are S-100 protein negative. The sustentacular cells are S-100 positive and focally positive for glial fibrillary acidic protein. By histochemistry, the paraganglioma cells are argyrophilic, periodic acid Schiff negative, mucicarmine negative, and argentaffin negative.[citation needed]
Sites of origin
[edit]About 85% of paragangliomas develop in the abdomen; only 12% develop in the chest and 3% in the head and neck region (the latter are the most likely to be symptomatic). While most are single, rare multiple cases occur (usually in a hereditary syndrome). [citation needed] Paragangliomas are described by their site of origin and are often given special names:
- Head and neck paraganglioma (HNPGL): There are various types of head and neck paraganglioma; they may have specialized names depending on the precise location.[9]
- Carotid paraganglioma (carotid body tumor): Is the most common of the head and neck paragangliomas. It usually presents as a painless neck mass, but larger tumors may cause cranial nerve palsies, usually of the vagus nerve and hypoglossal nerve.
- Glomus tympanicum and Glomus jugulare, also known as jugulotympanic paraganglioma: Both commonly present as a middle ear mass resulting in tinnitus (in 80%) and hearing loss (in 60%). The cranial nerves of the jugular foramen may be compressed, resulting swallowing difficulty, or ipsilateral weakness of the upper trapezius and sternocleiodomastoid muscles (from compression of the spinal accessory nerve). These patients present with a reddish bulge behind an intact ear drum. This condition is also known as the "Red drum". On application of pressure to the external ear canal with the help of a pneumatic ear speculum the mass could be seen to blanch. This sign is known as "Brown's sign". A deficient bony plate along the tympanic portion of the internal carotid artery (aberrant ICA) is a normal variant and can be mistaken with glomus jugulare.[10]
- Organ of Zuckerkandl: A collection of paraganglia near the bifurcation of the aorta, comprising a small mass of neural crest-derived chromaffin cells. Serves as a common origin of abdominal paragangliomas.
- Vagal paraganglioma: These are the least common of the head and neck paragangliomas. They usually present as a painless neck mass, but may result in dysphagia, hoarseness, or coughing with compression of the mass.
- Pulmonary paraganglioma: These occur in the lung and may be either single or multiple.[11]
- Other sites: Rare sites of involvement are the larynx, nasal cavity, paranasal sinuses, thyroid gland, and the thoracic inlet, as well as the bladder in extremely rare cases.
Diagnosis
[edit]Classification
[edit]Paragangliomas originate from paraganglia in chromaffin-negative glomus cells derived from the embryonic neural crest, functioning as part of the sympathetic nervous system (a branch of the autonomic nervous system). These cells normally act as special chemoreceptors located along blood vessels, particularly in the carotid bodies (at the bifurcation of the common carotid artery in the neck) and in aortic bodies (near the aortic arch).[citation needed]
Accordingly, paragangliomas are categorised as originating from a neural cell line in the World Health Organization classification of neuroendocrine tumors. In the categorization proposed by Wick, paragangliomas belong to group II.[12] Given the fact that they originate from cells of the orthosympathetic system, paragangliomas are closely related to pheochromocytomas, which however are chromaffin-positive.[citation needed]
Gallium-68 DOTATATE PET/CT imaging modality may be used to confirm the presence of a paraganglioma.[13]
Treatment
[edit]The main treatment modalities are surgery, embolization[14] and radiotherapy.[15] Treatment depends on a variety of factors, including patient symptoms, as well as tumor size and location.[16]
Additional images
[edit]-
 Micrograph of a carotid body tumor
Micrograph of a carotid body tumor -
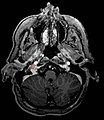
-
 Ectopic functional paraganglioma (glomus jugulare) in a patient with VHL. T2 weighted MRI at the same location demonstrates a high signal mass consistent with a paraganglioma. Extra adrenal paragangliomas can be found in VHL (arrow).
Ectopic functional paraganglioma (glomus jugulare) in a patient with VHL. T2 weighted MRI at the same location demonstrates a high signal mass consistent with a paraganglioma. Extra adrenal paragangliomas can be found in VHL (arrow). -
 S100 immunostain highlighting the sustentacular cells in a paraganglioma
S100 immunostain highlighting the sustentacular cells in a paraganglioma -
 Digital subtraction arteriogram of carotid body tumor and jugular paraganglioma
Digital subtraction arteriogram of carotid body tumor and jugular paraganglioma


_in_a_patient_with_VHL.jpg)


See also
[edit]References
[edit]- ^ Martins, Rute; Bugalho, Maria João (2014). "Paragangliomas/Pheochromocytomas: Clinically Oriented Genetic Testing". International Journal of Endocrinology. 2014: 794187. doi:10.1155/2014/794187. ISSN 1687-8337. PMC 4037125. PMID 24899893.
- ^ "Pheochromocytoma and Paraganglioma Treatment (PDQ®)–Patient Version - NCI". www.cancer.gov. December 23, 2011.
- ^ Elsamadicy, Emad A.; Yazdani, Shekoufeh; Karuppiah, Arunthevaraja; Marcano, Isabel; Turan, Ozhan; Kodali, Bhavani Shankar; Jessel, Rebecca (2021). "Paraganglioma Presenting as Hypoxia and Syncope in Pregnancy: A Case Report". A&A Practice. 15 (3): e01411. doi:10.1213/XAA.0000000000001411. PMID 33684077. S2CID 232159725.
- ^ Bayley JP, Kunst HP, Cascon A, Sampietro ML, Gaal J, Korpershoek E, Hinojar-Gutierrez A, Timmers HJ, Hoefsloot LH, Hermsen MA, Suárez C, Hussain AK, Vriends AH, Hes FJ, Jansen JC, Tops CM, Corssmit EP, de Knijff P, Lenders JW, Cremers CW, Devilee P, Dinjens WN, de Krijger RR, Robledo M (April 2010). "SDHAF2 mutations in familial and sporadic paraganglioma and phaeochromocytoma". The Lancet. Oncology. 11 (4): 366–72. doi:10.1016/S1470-2045(10)70007-3. PMID 20071235.
- ^ Qin Y, Yao L, King EE, Buddavarapu K, Lenci RE, Chocron ES, Lechleiter JD, Sass M, Aronin N, Schiavi F, Boaretto F, Opocher G, Toledo RA, Toledo SP, Stiles C, Aguiar RC, Dahia PL (March 2010). "Germline mutations in TMEM127 confer susceptibility to pheochromocytoma". Nature Genetics. 42 (3): 229–33. doi:10.1038/ng.533. PMC 2998199. PMID 20154675.
- ^ Comino-Méndez I, Gracia-Aznárez FJ, Schiavi F, Landa I, Leandro-García LJ, Letón R, Honrado E, Ramos-Medina R, Caronia D, Pita G, Gómez-Graña A, de Cubas AA, Inglada-Pérez L, Maliszewska A, Taschin E, Bobisse S, Pica G, Loli P, Hernández-Lavado R, Díaz JA, Gómez-Morales M, González-Neira A, Roncador G, Rodríguez-Antona C, Benítez J, Mannelli M, Opocher G, Robledo M, Cascón A (June 2011). "Exome sequencing identifies MAX mutations as a cause of hereditary pheochromocytoma". Nature Genetics. 43 (7): 663–7. doi:10.1038/ng.861. PMID 21685915. S2CID 205357831.
- ^ Buffet A, Morin A, Castro-Vega LJ, Habarou F, Lussey-Lepoutre C, Letouzé E, Lefebvre H, Guilhem I, Magalie H, Raingeard I, Padilla-Girola M, Tran T, Tchara L, Bertherat J, Amar L, Ottolenghi C, Burnichon N, Gimenez-Roqueplo AP, Favier J (February 2018). "Germline mutations in the mitochondrial 2-oxoglutarate/malate carrier SLC25A11 gene confer a predisposition to metastatic paragangliomas". Cancer Research. 78 (8): 1914–1922. doi:10.1158/0008-5472.CAN-17-2463. PMID 29431636.
- ^ Kairi-Vassilatou E, Argeitis J, Nika H, Grapsa D, Smyrniotis V, Kondi-Pafiti A (2007). "Malignant paraganglioma of the urinary bladder in a 44-year-old female: clinicopathological and immunohistochemical study of a rare entity and literature review". European Journal of Gynaecological Oncology. 28 (2): 149–51. PMID 17479683.
- ^ Sciacovelli, Marco; Schmidt, Christina; Maher, Eamonn R.; Frezza, Christian (2020). "Metabolic Drivers in Hereditary Cancer Syndromes". Annual Review of Cancer Biology. 4: 77–97. doi:10.1146/annurev-cancerbio-030419-033612.
- ^ Feky, Mostafa Mahmoud El. "Aberrant internal carotid artery | Radiology Case | Radiopaedia.org". radiopaedia.org. Retrieved 2017-05-02.
- ^ da Silva RA, Gross JL, Haddad FJ, Toledo CA, Younes RN (February 2006). "Primary pulmonary paraganglioma: case report and literature review". Clinics. 61 (1): 83–6. doi:10.1590/S1807-59322006000100015. PMID 16532231.
- ^ Wick MR (March 2000). "Neuroendocrine neoplasia. Current concepts". American Journal of Clinical Pathology. 113 (3): 331–5. doi:10.1309/ETJ3-QBUK-13QD-J8FP. PMID 10705811.
- ^ Chang, Chian A.; Pattison, David A.; Tothill, Richard W.; Kong, Grace; Akhurst, Tim J.; Hicks, Rodney J.; Hofman, Michael S. (2016-08-17). "68Ga-DOTATATE and 18F-FDG PET/CT in Paraganglioma and Pheochromocytoma: utility, patterns and heterogeneity". Cancer Imaging. 16 (1): 22. doi:10.1186/s40644-016-0084-2. ISSN 1740-5025. PMC 4989291. PMID 27535829.
- ^ Carlsen CS, Godballe C, Krogdahl AS, Edal AL (December 2003). "Malignant vagal paraganglioma: report of a case treated with embolization and surgery". Auris, Nasus, Larynx. 30 (4): 443–6. doi:10.1016/S0385-8146(03)00066-X. PMID 14656575.
- ^ Pitiakoudis M, Koukourakis M, Tsaroucha A, Manavis J, Polychronidis A, Simopoulos C (December 2004). "Malignant retroperitoneal paraganglioma treated with concurrent radiotherapy and chemotherapy". Clinical Oncology. 16 (8): 580–1. doi:10.1016/j.clon.2004.08.002. PMID 15630855.
- ^ "Paragangliomas / Glomus Tumors of the Head and Neck". Stanford Medicine. Retrieved 16 March 2023.
External links
[edit]- Sampath Chandra Prasad; Carlo Terenzio Paties; Mattia Russel Pantalone; Renato Mariani-Costantini; Mario Sanna (July 2, 2019). "Carotid Body and Vagal Paragangliomas: Epidemiology, Genetics, Clinicopathological Features, Imaging, and Surgical Management". In Mariani-Costantini, R (ed.). Paraganglioma: A Multidisciplinary Approach. Codon Publications. ISBN 9780994438171. PMID 31294944. Retrieved 16 March 2023.
- GeneReviews/NCBI/NIH/UW entry on Hereditary Paraganglioma-Pheochromocytoma Syndromes
| Authority control databases: National |
|---|