
Pontocerebellar hypoplasia
| Pontocerebellar hypoplasia | |
|---|---|
| Other names | Non-syndromic pontocerebellar hypoplasia |
 | |
| Pontocerebellar hypoplasia is inherited in an autosomal recessive manner | |
| Specialty | Neurology |
| Treatment | Unknown |
Pontocerebellar hypoplasia (PCH) is a heterogeneous group of rare neurodegenerative disorders caused by genetic mutations and characterised by progressive atrophy of various parts of the brain such as the cerebellum or brainstem (particularly the pons).[1] Where known, these disorders are inherited in an autosomal recessive fashion. There is no known cure for PCH.[2]
Signs and symptoms
There are different signs and symptoms for different forms of pontocerebellar hypoplasia, at least six of which have been described by researchers. All forms involve abnormal development of the brain, leading to slow development, movement problems, and intellectual impairment.[2]
-
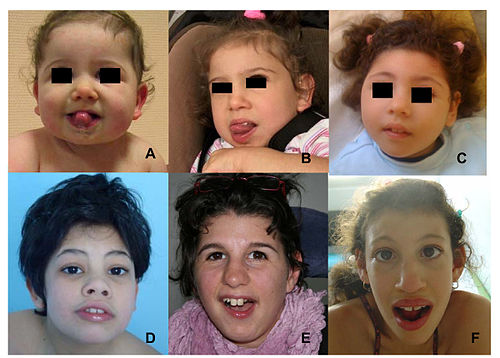 Facial features (dysmorphism) of patients with one form of pontocerebellar hypoplasia due to mutations in the CASK gene. A and B: patient at 1 year (A) and 4 years (B). C: patient, 18 months. D: patient, 13 years. E: patient, 13 years. F: patient, 12 years. Note minor facial dysmorphism: round face, small chin, well-drawn eyebrows in the younger patients; longer face, high and large nasal bridge, long nose, protruding maxilla, in the older patients.
Facial features (dysmorphism) of patients with one form of pontocerebellar hypoplasia due to mutations in the CASK gene. A and B: patient at 1 year (A) and 4 years (B). C: patient, 18 months. D: patient, 13 years. E: patient, 13 years. F: patient, 12 years. Note minor facial dysmorphism: round face, small chin, well-drawn eyebrows in the younger patients; longer face, high and large nasal bridge, long nose, protruding maxilla, in the older patients. -
 Magnetic resonance imaging (MRI) examples of patients with pontocerebellar hypoplasia with CASK mutations. A. Sagittal images showing different degrees of hypoplasia (incomplete formation) of the pons and vermis (parts of the brain). Numbers represent different patients. Figure 9a shows an MRI of a patient at age 4 months and figure 9b shows the same patient at age 11 years. There is no progression of the lesions between successive MRI in patient 9. Note that in all patients, the pons is very small but has a relative sparing of its bulging, mainly in its superior part. Hypoplasia predominates at the lower part of the pons. Vermis hypoplasia is very variable, severe in patient 13, very slight in patient 10-11-12 and also predominates at the inferior part. B. Coronal images showing varying degrees of cerebellar hemispheric (one of two halves of a part of the brain) hypoplasia. Hemispheres are frequently asymmetric. Note that the vermis does not protrude from the hemispheres indicating similar involvement of the vermis and the hemispheres. This pattern is different from that of PCH2 in which the vermis is relatively spared leading to the classic image of a "dragonfly", the protruding vermis being the body of the dragonfly and the hemispheres, the wings.
Magnetic resonance imaging (MRI) examples of patients with pontocerebellar hypoplasia with CASK mutations. A. Sagittal images showing different degrees of hypoplasia (incomplete formation) of the pons and vermis (parts of the brain). Numbers represent different patients. Figure 9a shows an MRI of a patient at age 4 months and figure 9b shows the same patient at age 11 years. There is no progression of the lesions between successive MRI in patient 9. Note that in all patients, the pons is very small but has a relative sparing of its bulging, mainly in its superior part. Hypoplasia predominates at the lower part of the pons. Vermis hypoplasia is very variable, severe in patient 13, very slight in patient 10-11-12 and also predominates at the inferior part. B. Coronal images showing varying degrees of cerebellar hemispheric (one of two halves of a part of the brain) hypoplasia. Hemispheres are frequently asymmetric. Note that the vermis does not protrude from the hemispheres indicating similar involvement of the vermis and the hemispheres. This pattern is different from that of PCH2 in which the vermis is relatively spared leading to the classic image of a "dragonfly", the protruding vermis being the body of the dragonfly and the hemispheres, the wings.


The following values seem to be aberrant in children with CASK gene defects: lactate, pyruvate, 2-ketoglutaric acid, adipic acid, and suberic acid which seems to support the thesis that CASK affects mitochondrial function.[3]
Causes
Pontocerebellar hypoplasia is caused by mutations in genes including Sepsecs gene, VRK1 (PCH1); TSEN2, TSEN34 (PCH2); RARS2 (PCH6); and TSEN54 (PCH2 and PCH4). The genes associated with PCH3 and PCH5 have not yet been identified.[2]
The mutated genes in PCH are autosomal recessive, which means that parents of an affected child each carry only one copy of the damaged gene. In each parent the other copy performs its proper function and they display no signs of PCH. A child inheriting two damaged copies of the gene will be affected by PCH.[2]
Mechanism
Mutations in the genes that cause PCH produce faults in the production of chemicals, usually enzymes, that are required for the development of nerve cells (neurons) and for properly processing RNA, which is needed for any cell to function normally. The exact mechanism by which PCH affects the development of the cerebellum and pons is not well understood.[2]
Diagnosis
Classification
Pontocerebellar hypoplasia is classified as follows:[4]
| Type | OMIM | Gene | Locus | Distinctive features | Alternate names |
|---|---|---|---|---|---|
| PCH1A | 607596 | VRK1 | 14q32 | Infantile onset anterior horn cell degeneration resulting in progressive muscle atrophy; resembles infantile spinal muscular atrophy[5] | Spinal muscular atrophy with pontocerebellar hypoplasia (SMA-PCH) |
| PCH1B | 614678 | EXOSC3 | 9p13.2 | Cerebellar and spinal motor neuron degeneration beginning at birth and resulting in decreased body tone, respiratory insufficiency, muscle atrophy, progressive microcephaly and global developmental delay[6] | |
| PCH2A | 277470 | TSEN54 | 17q25.1 | Dyskinetic movements, seizures (frequently) | Volendam neurodegenerative disease |
| PCH2B | 612389 | TSEN2 | 3p25.2 | ||
| PCH2C | 612390 | TSEN34 | 19q13.42 | ||
| PCH2D | 613811 | SEPSECS | 4p15.2 | Progressive cerebello-cerebral atrophy (PCCA) | |
| PCH2E | 615851 | VPS53 | 17p13.3 | Profound mental retardation, progressive microcephaly, spasticity, and early-onset epilepsy[7] | |
| PCH2F | 617026 | TSEN15 | 1q25.3 | Variable neurologic signs and symptoms, including cognitive and motor delay, poor or absent speech, seizures, and spasticity | |
| PCH3 | 608027 | PCLO | 7q11–q21 | Seizures, short stature, optic atrophy, progressive microcephaly, severe developmental delay; described only in a handful of cases.[8] | CLAM-PCH, cerebellar atrophy with progressive microcephaly |
| PCH4 | 225753 | TSEN54 | 17q25.1 | Severe prenatal form of PCH2 with excess fluid in the amniotic sac, muscle contractures, brief involuntary muscle twitching, brief episodes without breathing, and early death following birth | |
| PCH5 | 610204 | TSEN54 | 17q25.1 | Severe prenatal form, described in one family | Olivopontocerebellar hypoplasia (OPCH) |
| PCH6 | 611523 | RARS2 | 6q15 | Severe encephalopathy in the newborn with hypotonia, and inconstantly: intractable seizures, edema, increased lactate blood levels, mitochondrial respiratory chain defects | |
| PCH7 | 614969 | TOE1 | 1p34.1 | Hypotonia, apneic episodes, seizures, vanishing testis[9][10] | |
| PCH8 | 614961 | CHMP1A | 16q24.3 | Severe psychomotor retardation, abnormal movements, hypotonia, spasticity, and variable visual defects[11] | |
| PCH9 | 615809 | AMPD2 | 1p13.3 | Severely delayed psychomotor development, progressive microcephaly, spasticity, seizures, and brain abnormalities, including brain atrophy, thin corpus callosum, and delayed myelination[12] | |
| PCH10 | 615803 | CLP1 | 11q12.1 | Severely delayed psychomotor development, progressive microcephaly, spasticity, seizures, and brain abnormalities, including brain atrophy and delayed myelination[13] |
Pontine and cerebellar hypoplasia is also observed in certain phenotypes of X-linked mental retardation – so called MICPCH.
Another gene that has been associated with this condition is coenzyme A synthase (COASY).[14]
Treatment
This section is empty. You can help by adding to it. (December 2017) |
Outcomes
The severity of different forms of PCH varies, but many children inheriting the mutated gene responsible do not survive infancy[15] or childhood; nevertheless, some individuals born with PCH have reached adulthood.[2]
See also
References
- ^ Millen KJ, Gleeson JG (February 2008). "Cerebellar development and disease". Curr Opin Neurobiol. 18 (1): 12–9. doi:10.1016/j.conb.2008.05.010. PMC 2474776. PMID 18513948.
- ^ a b c d e f "Pontocerebellar hypoplasia". Genetics Home Reference. U.S. National Library of Medicine. December 2009. Retrieved 20 September 2014.
- ^ Mukherjee, K; Slawson, JB; Christmann, BL; Griffith, LC (2014). "Neuron-specific protein interactions of Drosophila CASK-β are revealed by mass spectrometry". Frontiers in Molecular Neuroscience. 7: 58. doi:10.3389/fnmol.2014.00058. PMC 4075472. PMID 25071438.
- ^ Online Mendelian Inheritance in Man (OMIM): [1]
- ^ Online Mendelian Inheritance in Man (OMIM): 607596
- ^ Online Mendelian Inheritance in Man (OMIM): 614678
- ^ Online Mendelian Inheritance in Man (OMIM): 615851
- ^ Online Mendelian Inheritance in Man (OMIM): 608027
- ^ Anderson, C; Davies, JH; Lamont, L; Foulds, N (April 2011). "Early pontocerebellar hypoplasia with vanishing testes: A new syndrome?". American Journal of Medical Genetics Part A. 155A (4): 667–72. doi:10.1002/ajmg.a.33897. PMID 21594990. S2CID 37977323.
- ^ Namavar, Y; Barth, PG; Poll-The, BT; Baas, F (2011). "Classification, diagnosis and potential mechanisms in pontocerebellar hypoplasia". Orphanet Journal of Rare Diseases. 6: 50. doi:10.1186/1750-1172-6-50. PMC 3159098. PMID 21749694.
- ^ Online Mendelian Inheritance in Man (OMIM): 614961
- ^ Online Mendelian Inheritance in Man (OMIM): 615809
- ^ Online Mendelian Inheritance in Man (OMIM): 615803
- ^ van Dijk T, Ferdinandusse S, Ruiter JPN, Alders M, Mathijssen IB, Parboosingh JS, Innes AM, Meijers-Heijboer H, Poll-The BT, Bernier FP, Wanders RJA, Lamont RE, Baas F (2018) Biallelic loss of function variants in COASY cause prenatal onset pontocerebellar hypoplasia, microcephaly, and arthrogryposis. Eur J Hum Genet doi: 10.1038/s41431-018-0233-0
- ^ Basson MA, Wingate RJ (September 2013). "Congenital hypoplasia of the cerebellum: developmental causes and behavioral consequences". Front Neuroanat. 7: 29. doi:10.3389/fnana.2013.00029. PMC 3759752. PMID 24027500.