
Immune thrombocytopenic purpura
| Immune thrombocytopenic purpura | |
|---|---|
| Other names | Idiopathic thrombocytopenic purpura, idiopathic immune thrombocytopenia, primary immune thrombocytopenia, idiopathic thrombocytopenic purpura, primary immune thrombocytopenic purpura, autoimmune thrombocytopenic purpura |
 | |
| Petechiae, or small bruise-like markings, may occur in ITP | |
| Specialty | Hematology |
| Frequency | 50–100 per million (12-month risk) |
Immune thrombocytopenic purpura (ITP), also known as idiopathic thrombocytopenic purpura or immune thrombocytopenia, is an autoimmune primary disorder of hemostasis characterized by a low platelet count in the absence of other causes.[1][2] ITP often results in an increased risk of bleeding from mucosal surfaces (such as the nose or gums) or the skin (causing purpura and bruises).[1] Depending on which age group is affected, ITP causes two distinct clinical syndromes: an acute form observed in children and a chronic form in adults. Acute ITP often follows a viral infection and is typically self-limited (resolving within two months), while the more chronic form (persisting for longer than six months) does not yet have a specific identified cause. Nevertheless, the pathogenesis of ITP is similar in both syndromes involving antibodies against various platelet surface antigens such as glycoproteins.
Diagnosis of ITP involves identifying a low platelet count through a complete blood count, a common blood test. However, since the diagnosis relies on excluding other potential causes of a low platelet count, additional investigations, such as a bone marrow biopsy, may be necessary in certain cases.
For mild cases, careful observation may be sufficient. However, in instances of very low platelet counts or significant bleeding, treatment options may include corticosteroids, intravenous immunoglobulin, anti-D immunoglobulin, or immunosuppressive medications. Refractory ITP, which does not respond to conventional treatment or shows constant relapse after splenectomy, requires treatment to reduce the risk of significant bleeding.[3] Platelet transfusions may be used in severe cases with extremely low platelet counts in individuals experiencing bleeding. In some cases, the body may compensate by producing abnormally large platelets.
Signs and symptoms
[edit]Signs of ITP include the spontaneous formation of bruises (purpura) and petechiae (tiny bruises), especially on the extremities. Additionally, bleeding from the nostrils and/or gums, as well as menorrhagia (excessive menstrual bleeding), may occur if the platelet count falls below 20,000 per μL.[4] A platelet count below 10,000 per μL can lead to the spontaneous formation of hematomas (blood masses) in the mouth or on other mucous membranes. Furthermore, bleeding time from minor lacerations or abrasions is usually prolonged.[citation needed]
In cases where platelet counts drop to extremely low levels (<5,000 per μL), serious and potentially fatal complications may arise. These complications include subarachnoid or intracerebral hemorrhage (bleeding inside the skull or brain), lower gastrointestinal bleeding, or other internal bleeding. A person with ITP with an extremely low platelet count is susceptible to internal bleeding resulting from blunt abdominal trauma, such as in a motor vehicle crash. These complications are more likely to occur when the platelet count is less than 20,000 per μL.[5]
-
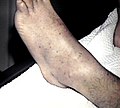 Petechiae on the lower extremities
Petechiae on the lower extremities -
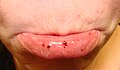 Oral petechiae/purpura — lower lip
Oral petechiae/purpura — lower lip -
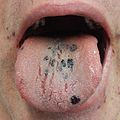 Petechia on the tongue in a person with platelets of 3 due to ITP
Petechia on the tongue in a person with platelets of 3 due to ITP -
 Petechia of the lower leg in a person with platelets of 3 due to ITP
Petechia of the lower leg in a person with platelets of 3 due to ITP




Pathogenesis
[edit]In approximately 60 percent of cases, antibodies against platelets can be detected.[6] Most often these antibodies are against platelet membrane glycoproteins IIb-IIIa or Ib-IX, and are of the immunoglobulin G (IgG) type. The Harrington–Hollingsworth experiment established the immune pathogenesis of ITP.[7]
The coating of platelets with IgG renders them susceptible to opsonization and phagocytosis by splenic macrophages, as well by Kupffer cells in the liver. The IgG autoantibodies are also thought to damage megakaryocytes, the precursor cells to platelets, although this is believed to contribute only slightly to the decrease in platelet numbers. Recent research now indicates that impaired production of the glycoprotein hormone, thrombopoietin, which is the stimulant for platelet production, may be a contributing factor to the reduction in circulating platelets. This observation has led to the development of a class of ITP-targeted medications referred to as thrombopoietin receptor agonists.[5]
The stimulus for auto-antibody production in ITP is probably abnormal T cell activity.[8][9][10] Preliminary findings suggest that these T cells can be influenced by medications that target B cells, such as rituximab.[11]
Diagnosis
[edit]
The diagnosis of ITP is a process of exclusion. First, it has to be determined that there are no blood abnormalities other than a low platelet count, and no physical signs other than bleeding. Then, secondary causes (around 5–10 percent of suspected ITP cases) should be excluded including medications (quinine or heparin), viral infection (HIV or HCV), malignancy (leukemia), autoimmune conditions (systemic lupus erythematosus or antiphospholipid syndrome), onyalai, and others.[4][12] All patients with presumed ITP should be tested for HIV and hepatitis C virus, as platelet counts may be corrected by treating the underlying disease. In approximately 2.7 to 5 percent of cases, autoimmune hemolytic anemia and ITP coexist, a condition referred to as Evans syndrome.[13][14]
Despite the destruction of platelets by splenic macrophages, the spleen is normally not enlarged. In fact, an enlarged spleen should lead to a search for other possible causes for the thrombocytopenia. Bleeding time is usually prolonged in ITP patients. However, the use of bleeding time in diagnosis is discouraged by the American Society of Hematology practice guidelines[15] and a normal bleeding time does not exclude a platelet disorder.[16]
Bone marrow examination may be performed on patients over the age of 60 and those who do not respond to treatment, or when the diagnosis is in doubt.[12] On examination of the marrow, an increase in the production of megakaryocytes may be observed and may help in establishing a diagnosis of ITP. An analysis for anti-platelet antibodies is a matter of clinician's preference, as there is disagreement on whether the 80 percent specificity of this test is sufficient to be clinically useful.[12]
Treatment
[edit]With rare exceptions, there is usually no need to treat based on platelet counts. Many older recommendations suggested a certain platelet count threshold (usually somewhere below 20.0/nL) as an indication for hospitalization or treatment. Current guidelines recommend treatment for adults with significant bleeding or counts below 30/nL, with very low certainty of evidence.[17] Treatment recommendations sometimes differ for adult and pediatric ITP.[18]
Steroids
[edit]Initial treatment usually consists of the administration of corticosteroids, a group of medications that suppress the immune system. The dose and mode of administration is determined by platelet count and whether there is active bleeding: in urgent situations, infusions of dexamethasone or methylprednisolone may be used, while oral prednisone or prednisolone may suffice in less severe cases. Once the platelet count has improved, the dose of steroid is gradually reduced while the possibility of relapse is monitored. 60–90 percent will experience a relapse during dose reduction or cessation.[12][19] Long-term steroids are avoided if possible because of potential side-effects that include osteoporosis, diabetes and cataracts.[20]
Anti-D
[edit]Another option, suitable for Rh-positive patients with functional spleens is intravenous administration of Rho(D) immune globulin [Human; Anti-D]. The mechanism of action of anti-D is not fully understood. However, following administration, anti-D-coated red blood cell complexes saturate Fcγ receptor sites on macrophages, resulting in preferential destruction of red blood cells (RBCs), therefore sparing antibody-coated platelets. There are two anti-D products indicated for use in patients with ITP: WinRho SDF and Rhophylac. The most common adverse reactions are headache (15%), nausea/vomiting (12%) chills (<2%) and fever (1%).[citation needed]. Following a black-box warning of possible uncontrolled hemolytic reactions, use of intravenous anti-D declined sharply.[21] Intramuscular anti-D has been suggested as an alternative, with one case series reporting a 73% response rate.[22]
Steroid-sparing agents
[edit]There is increasing use of immunosuppressants such as mycophenolate mofetil and azathioprine because of their effectiveness. In chronic refractory cases, where immune pathogenesis has been confirmed,[23] the off-label use of the vinca alkaloid[24][25][26] and chemotherapy agent vincristine may be attempted.[27][28] However, vincristine has significant side effects[29] and its use in treating ITP must be approached with caution, especially in children.
Intravenous immunoglobulin
[edit]Intravenous immunoglobulin (IVIg) may be infused in some cases in order to decrease the rate at which macrophages consume antibody-tagged platelets. However, while sometimes effective, it is costly and produces improvement that generally lasts less than a month. Nevertheless, in the case of an ITP patient already scheduled for surgery who has a dangerously low platelet count and has experienced a poor response to other treatments, IVIg can rapidly increase platelet counts, and can also help reduce the risk of major bleeding by transiently increasing platelet counts.[30]
Thrombopoietin receptor agonists
[edit]Thrombopoietin receptor agonists are pharmaceutical agents that stimulate platelet production in the bone marrow. In this, they differ from the previously discussed agents that act by attempting to curtail platelet destruction.[31] Two such products are currently available:
- Romiplostim (trade name Nplate) is a thrombopoiesis stimulating Fc-peptide fusion protein (peptibody) that is administered by subcutaneous injection. Designated an orphan drug in 2003 under United States law, clinical trials demonstrated romiplostim to be effective in treating chronic ITP, especially in relapsed post-splenectomy patients.[32][33] Romiplostim was approved by the United States Food and Drug Administration (FDA) for long-term treatment of adult chronic ITP on August 22, 2008.[34]
- Eltrombopag (trade name Promacta in the US, Revolade in the EU) is an orally-administered agent with an effect similar to that of romiplostim. It too has been demonstrated to increase platelet counts and decrease bleeding in a dose-dependent manner.[35] Developed by GlaxoSmithKline and also designated an orphan drug by the FDA, Promacta was approved by the FDA on November 20, 2008.[36]
Thrombopoietin receptor agonists exhibited the greatest success so far in treating patients with refractory ITP.[37]
Side effects of thrombopoietin receptor agonists include headache, joint or muscle pain, dizziness, nausea or vomiting, and an increased risk of blood clots.[31]
Surgery
[edit]Splenectomy (removal of the spleen) may be considered in patients who are either unresponsive to steroid treatment, have frequent relapses, or cannot be tapered off steroids after a few months. Platelets which have been bound by antibodies are taken up by macrophages in the spleen (which have Fc receptors), and so removal of the spleen reduces platelet destruction. The procedure is potentially risky in ITP cases due to the increased possibility of significant bleeding during surgery. Durable remission following splenectomy is achieved in 60 — 80 percent of ITP cases.[38] Even though there is a consensus regarding the short-term efficacy of splenectomy, findings on its long-term efficacy and side-effects are controversial.[37][39] After splenectomy, 11.6–75 percent of ITP cases relapsed, and 8.7–40 percent of ITP cases had no response to splenectomy.[37][40][41][42] The use of splenectomy to treat ITP has diminished since the development of steroid therapy and other pharmaceutical remedies.[43]
Platelet transfusion
[edit]Platelet transfusion alone is normally not recommended except in an emergency and is usually unsuccessful in producing a long-term platelet count increase. This is because the underlying autoimmune mechanism that is destroying the patient's platelets will also destroy donor platelets, and so platelet transfusions are not considered a long-term treatment option.[44]
H. pylori eradication
[edit]In adults, particularly those living in areas with a high prevalence of Helicobacter pylori (which normally inhabits the stomach wall and has been associated with peptic ulcers), identification and treatment of this infection has been shown to improve platelet counts in a third of patients. In a fifth, the platelet count normalized completely; this response rate is similar to that found in treatment with rituximab, which is more expensive and less safe.[45] In children, this approach is not supported by evidence, except in high prevalence areas. Urea breath testing and stool antigen testing perform better than serology-based tests; moreover, serology may be false-positive after treatment with IVIG.[46]
Other agents
[edit]- Dapsone (also called diphenylsulfone, DDS, or avlosulfon) is an anti-infective sulfone medication. Dapsone may also be helpful in treating lupus, rheumatoid arthritis, and as a second-line treatment for ITP. The mechanism by which dapsone assists in ITP is unclear but an increased platelet count is seen in 40–60 percent of recipients.[47][48]
- The off-label use of rituximab, a chimeric monoclonal antibody against the B cell surface antigen CD20, may sometimes be an effective alternative to splenectomy. However, significant side-effects can occur, and randomized controlled trials are inconclusive.[49]
- Fostamatinib was approved for refractory chronic ITP in the United States in April 2018.[50] Fostamatinib blocks the activity of the enzyme spleen tyrosine kinase (SYK), thus reducing FcγR-mediated signal transduction and phagocytosis by monocyte/macrophages. Adverse effects include diarrhea, hypertension, and abnormal liver function tests.[51]
- In a phase 1-2 open-label study treatment with CM313, a novel anti-CD38 monoclonal antibody, rapidly boosted platelet levels in adults with ITP by inhibiting antibody-dependent cell-mediated cytotoxicity on platelets; maintained long-term efficacy by clearing plasma cells; and was associated with low-grade toxic effects.[52]
- Ianalumab is a monoclonal antibody that targets the BAFF receptor BAFF-R to inhibit B-cell signaling and deplete B cells through antibody-dependent cellular cytotoxicity. The VAYHIT1 study was designed to evaluate the efficacy and safety of ianalumab for ITP.[53]
Prognosis
[edit]In general, patients with acute ITP will only rarely have life-threatening bleeding.[54] Most patients ultimately have lower, but stable platelet counts, which are still hemostatic for the patient. Unlike children and adolescents, ITP is often chronic in adults, even after a splenectomy.[40]
Epidemiology
[edit]A normal platelet count is considered to be in the range of 150,000–450,000 per microlitre (μL) of blood for most healthy individuals. Hence one may be considered thrombocytopenic below that range, although the threshold for a diagnosis of ITP is not tied to any specific number.[2]
The incidence of ITP is estimated at 50–100 new cases per million per year, with children accounting for half of that number. At least 70 percent of childhood cases will end up in remission within six months, even without treatment.[55][56][57] Moreover, a third of the remaining chronic cases will usually remit during follow-up observation, and another third will end up with only mild thrombocytopenia (defined as a platelet count above 50,000).[55] A number of immune related genes and polymorphisms have been identified as influencing predisposition to ITP, with FCGR3a-V158 allele and KIRDS2/DL2 increasing susceptibility and KIR2DS5 shown to be protective.[58][59]
ITP is usually chronic in adults[60] and the probability of durable remission is 20–40 percent.[19] The male to female ratio in the adult group varies from 1:1.2 to 1.7 in most age ranges (childhood cases are roughly equal for both sexes) and the median age of adults at the diagnosis is 56–60.[12] The ratio between male and female adult cases tends to widen with age. In the United States, the adult chronic population is thought to be approximately 60,000—with women outnumbering men approximately 2 to 1, which has resulted in ITP being designated an orphan disease.[61]
The mortality rate due to chronic ITP varies but tends to be higher relative to the general population for any age range. In a study conducted in Great Britain, it was noted that ITP causes an approximately 60 percent higher rate of mortality compared to sex- and age-matched subjects without ITP. This increased risk of death with ITP is largely concentrated in the middle-aged and elderly. Ninety-six percent of reported ITP-related deaths were individuals 45 years or older. No significant difference was noted in the rate of survival between males and females.[62]
Pregnancy
[edit]The incidence of ITP in pregnancy is not well known. It may occur during any trimester of pregnancy. It is the most common cause of significant thrombocytopenia (platelets less than 100,000) in the second trimester, and it is a common cause of significant thrombocytopenia in the first and third trimesters.[63] As in non-pregnant individuals, ITP in pregnancy is a diagnosis of exclusion and other potential causes of low platelets in pregnancy require consideration. These include obstetrical causes such as pre-eclampsia, HELLP syndrome (hemolysis, elevated liver enzymes and low platelets), or thrombotic microangiopathies that may occur during pregnancy.[63] Other causes of thrombocytopenia which may occur in pregnancy, such as drug induced thrombocytopenia, hereditary thrombocytopenia and pseudothrombocytopenia should also be ruled out.[63] ITP can be difficult to distinguish from gestational thrombocytopenia (which is by far the most common cause of thrombocytopenia in pregnancy). Unlike ITP, the platelet count in gestational thrombocytopenia rarely goes below 100,000, and a platelet count below 80,000 is even more rare (seen in less than 0.1% of cases of gestational thrombocytopenia). Also unlike ITP, gestational thrombocytopenia is not a cause of neonatal or maternal bleeding, or neonatal thrombocytopenia.[63]
Women with ITP often have a decrease in their platelet counts when they become pregnant, often requiring treatment.[63] Pregnant women with ITP are 1.83 times more likely to have bleeding episodes during pregnancy compared to non-pregnant females with ITP, however, with proper treatment, platelets rarely drop below 30,000.[63] In ITP, severe bleeding is a rare occurrence, and with treatment maternal deaths due to ITP are extremely rare.[63] ITP has not been found to increase the risk of some common obstetrical complications; with no increased risk of pre-eclampsia, premature delivery, placental abruption or blood clots observed. Further, in those with ITP, platelet counts usually return to pre-pregnancy levels after delivery.[63]
Anti-platelet autoantibodies in a pregnant woman with ITP will attack the patient's own platelets and will also cross the placenta and react against fetal platelets leading to thrombocytopenia. Thrombopoietin levels are also increased during pregnancy (which is thought to be due to placental production of estradiol) and this estradiol leads to decreased megakaryocyte activity and therefore decreased platelet production.[63] Therefore, ITP is a significant cause of fetal and neonatal immune thrombocytopenia. Approximately 10% of newborns affected by ITP will have platelet counts <50,000/uL and 1% to 2% will have a risk of intracerebral hemorrhage, comparable to that of infants with neonatal alloimmune thrombocytopenia (NAIT).[64][65]
No lab test can reliably predict if neonatal thrombocytopenia will occur. The risk of neonatal thrombocytopenia is increased with:[66]
- Mothers with a history of splenectomy for ITP
- Mothers who had a previous infant affected with ITP
- Gestational (maternal) platelet count less than 100,000/uL
It is recommended that pregnant women with thrombocytopenia or a previous diagnosis of ITP be tested for serum antiplatelet antibodies. Indications for treatment of pregnant people with ITP include the presence of bleeding, platelet counts less than 20-30,000, planned procedures (such as an amniocentesis), and raising platelet levels prior to delivery (the minimum platelet level for a vaginal delivery is 30,000 and for a cesarean section is 50,000).[63] Rarely, a platelet transfusion may be needed to facilitate safe delivery.[63] First line therapies for ITP include steroids such as prednisone, IVIG or both. Second line therapies (usually used in those in whom first line therapies fail and thrombocytopenia persists) include rituximab, thrombopoetin receptor agonsists (such as romiplostim), or a splenectomy.[63] Platelet transfusions may be performed in newborns, depending on the degree of thrombocytopenia. IVIG and steroids (methylprednisolone) may also be given to raise the platelet counts.[63] It is recommended that neonates be followed with serial platelet counts for the first few days after birth.[64][66] Ultrasound or MRI are also recommended for severely thrombocytopenic infants to rule out a brain bleed.[63]
History
[edit]After initial reports by the Portuguese physician Amato Lusitano in 1556 and Lazarus de la Rivière (physician to the King of France) in 1658, it was the German physician and poet Paul Gottlieb Werlhof who in 1735 wrote the most complete initial report of the purpura of ITP. Platelets were unknown at the time.[67] The name "Werlhof's disease" was used more widely before the current descriptive name became more popular.[67][68] Platelets were described in the early 19th century, and in the 1880s several investigators linked the purpura with abnormalities in the platelet count.[67][69] The first report of a successful therapy for ITP was in 1916, when a young Polish medical student, Paul Kaznelson, described a female patient's response to a splenectomy.[67] Splenectomy remained a first-line remedy until the introduction of steroid therapy in the 1950s.[67]
References
[edit]- ^ a b Pietras, Nicole M.; Pearson-Shaver, Anthony L. (2022). "Immune Thrombocytopenic Purpura". StatPearls. StatPearls Publishing. PMID 32965953.
- ^ a b Rodeghiero F, Stasi R, Gernsheimer T, Michel M, Provan D, Arnold DM, et al. (March 2009). "Standardization of terminology, definitions and outcome criteria in immune thrombocytopenic purpura of adults and children: report from an international working group". Blood. 113 (11): 2386–93. doi:10.1182/blood-2008-07-162503. PMID 19005182.
- ^ Lambert MP, Gernsheimer TB (May 2017). "Clinical updates in adult immune thrombocytopenia". Blood. 129 (21): 2829–35. doi:10.1182/blood-2017-03-754119. PMC 5813736. PMID 28416506.
- ^ a b Cines DB, McMillan R (2005). "Management of adult idiopathic thrombocytopenic purpura". Annual Review of Medicine. 56: 425–42. doi:10.1146/annurev.med.56.082103.104644. PMID 15660520.
- ^ a b "Immune Thrombocytopenic Purpura". The Lecturio Medical Concept Library. Retrieved 27 July 2021.
- ^ Coopamah MD, Garvey MB, Freedman J, Semple JW (January 2003). "Cellular immune mechanisms in autoimmune thrombocytopenic purpura: An update". Transfusion Medicine Reviews. 17 (1): 69–80. doi:10.1053/tmrv.2003.50004. PMID 12522773.
- ^ Schwartz RS (November 2007). "Immune thrombocytopenic purpura--from agony to agonist". The New England Journal of Medicine. 357 (22): 2299–301. doi:10.1056/NEJMe0707126. PMID 18046034. S2CID 9765360.
- ^ Semple JW, Freedman J (November 1991). "Increased antiplatelet T helper lymphocyte reactivity in patients with autoimmune thrombocytopenia". Blood. 78 (10): 2619–25. doi:10.1182/blood.V78.10.2619.2619. PMID 1840468.
- ^ Stasi R, Cooper N, Del Poeta G, Stipa E, Laura Evangelista M, Abruzzese E, Amadori S (August 2008). "Analysis of regulatory T-cell changes in patients with idiopathic thrombocytopenic purpura receiving B cell-depleting therapy with rituximab". Blood. 112 (4): 1147–50. doi:10.1182/blood-2007-12-129262. PMID 18375792. S2CID 206867628.
- ^ Yu J, Heck S, Patel V, Levan J, Yu Y, Bussel JB, Yazdanbakhsh K (August 2008). "Defective circulating CD25 regulatory T cells in patients with chronic immune thrombocytopenic purpura". Blood. 112 (4): 1325–8. doi:10.1182/blood-2008-01-135335. PMC 2515134. PMID 18420827.
- ^ Godeau B, Porcher R, Fain O, Lefrère F, Fenaux P, Cheze S, et al. (August 2008). "Rituximab efficacy and safety in adult splenectomy candidates with chronic immune thrombocytopenic purpura: results of a prospective multicenter phase 2 study". Blood. 112 (4): 999–1004. doi:10.1182/blood-2008-01-131029. PMID 18463354.
- ^ a b c d e Cines DB, Bussel JB (October 2005). "How I treat idiopathic thrombocytopenic purpura (ITP)". Blood. 106 (7): 2244–51. doi:10.1182/blood-2004-12-4598. PMID 15941913.
- ^ Hansen DL, Möller S, Andersen K, Gaist D, Frederiksen H (October 2019). "Evans syndrome in adults — incidence, prevalence, and survival in a nationwide cohort". American Journal of Hematology. 94 (10): 1081–90. doi:10.1002/ajh.25574. PMID 31292991. S2CID 195879785.
- ^ Michel M, Chanet V, Dechartres A, Morin AS, Piette JC, Cirasino L, et al. (October 2009). "The spectrum of Evans syndrome in adults: new insight into the disease based on the analysis of 68 cases". Blood. 114 (15): 3167–72. doi:10.1182/blood-2009-04-215368. PMID 19638626.
- ^ "Diagnosis and treatment of idiopathic thrombocytopenic purpura: recommendations of the American Society of Hematology. The American Society of Hematology ITP Practice Guideline Panel". Annals of Internal Medicine. 126 (4): 319–26. February 1997. doi:10.7326/0003-4819-126-4-199702150-00010. PMID 9036806. S2CID 53089071.
- ^ Liesner RJ, Machin SJ (March 1997). "ABC of clinical haematology. Platelet disorders". BMJ. 314 (7083): 809–12. doi:10.1136/bmj.314.7083.809. PMC 2126215. PMID 9081003.
- ^ Neunert C, Terrell DR, Arnold DM, Buchanan G, Cines DB, Cooper N, Cuker A, Despotovic JM, George JN, Grace RF, Kühne T, Kuter DJ, Lim W, McCrae KR, Pruitt B, Shimanek H, Vesely SK (December 2019). "American Society of Hematology 2019 guidelines for immune thrombocytopenia". Blood Adv. 3 (23): 3829–66. doi:10.1182/bloodadvances.2019000966. PMC 6963252. PMID 31794604.
- ^ Neunert C, Lim W, Crowther M, Cohen A, Solberg L, Crowther MA (April 2011). "The American Society of Hematology 2011 evidence-based practice guideline for immune thrombocytopenia". Blood. 117 (16): 4190–207. doi:10.1182/blood-2010-08-302984. PMID 21325604.
- ^ a b Stevens W, Koene H, Zwaginga JJ, Vreugdenhil G (November 2006). "Chronic idiopathic thrombocytopenic purpura: present strategy, guidelines and new insights". The Netherlands Journal of Medicine. 64 (10): 356–63. PMID 17122451.
- ^ Buchman AL (October 2001). "Side effects of corticosteroid therapy". Journal of Clinical Gastroenterology. 33 (4): 289–94. doi:10.1097/00004836-200110000-00006. PMID 11588541. S2CID 23828538.
- ^ Bussel JB (February 2023). "Why should intramuscular anti-D be different from intravenous anti-D?". Br J Haematol. 200 (3): 275–6. doi:10.1111/bjh.18524. PMID 36408739.
- ^ Lakhwani S, López-Las Heras A, Rodríguez-García P, Iraheta S, Martín-Santos T, Rodríguez-Salazar MJ, Machado P, Hernández MT (February 2023). "Intramuscular Anti-D treatment for immune thrombocytopenia: A single centre experience". Br J Haematol. 200 (3): 353–7. doi:10.1111/bjh.18484. PMC 10091692. PMID 36198407.
- ^ Psaila B, Bussel JB (October 2008). "Refractory immune thrombocytopenic purpura: current strategies for investigation and management". British Journal of Haematology. 143 (1): 16–26. doi:10.1111/j.1365-2141.2008.07275.x. PMID 18573111. S2CID 38433249.
- ^ van Der Heijden R, Jacobs DI, Snoeijer W, Hallard D, Verpoorte R (March 2004). "The Catharanthus alkaloids: pharmacognosy and biotechnology". Current Medicinal Chemistry. 11 (5): 607–28. doi:10.2174/0929867043455846. PMID 15032608.
- ^ Raviña E (2011). "Vinca alkaloids". The evolution of drug discovery: From traditional medicines to modern drugs. Wiley. pp. 157–9. ISBN 978-3-527-32669-3. OCLC 1310590907.
- ^ Cooper R, Deakin JJ (2016). "Africa's gift to the world". Botanical Miracles: Chemistry of Plants That Changed the World. CRC Press. pp. 46–51. ISBN 978-1-4987-0430-4.
- ^ Kuboyama T, Yokoshima S, Tokuyama H, Fukuyama T (August 2004). "Stereocontrolled total synthesis of (+)-vincristine". Proceedings of the National Academy of Sciences of the United States of America. 101 (33): 11966–70. doi:10.1073/pnas.0401323101. PMC 514417. PMID 15141084.
- ^ Keglevich P, Hazai L, Kalaus G, Szántay C (May 2012). "Modifications on the basic skeletons of vinblastine and vincristine". Molecules. 17 (5): 5893–914. doi:10.3390/molecules17055893. PMC 6268133. PMID 22609781.
- ^ Ishida Y, Tomiyama Y, eds. (2017). Autoimmune Thrombocytopenia. Springer Nature. ISBN 978-981-10-4142-6.
- ^ Godeau, Bertrand; Caulier, Marie-Thérèse; Decuypere, Laurent; Rose, Christian; Schaeffer, Annette; Bierling, Philippe (1999). "Intravenous immunoglobulin for adults with autoimmune thrombocytopenic purpura: results of a randomized trial comparing 0.5 and 1 g/kg b.w." British Journal of Haematology. 107 (4): 716–9. doi:10.1046/j.1365-2141.1999.01766.x. ISSN 1365-2141. PMID 10606875. S2CID 27193777.
- ^ a b "Idiopathic thrombocytopenic purpura: Treatments and Drugs". Mayo Clinic. Retrieved October 16, 2012.
- ^ Bussel JB, Kuter DJ, George JN, McMillan R, Aledort LM, Conklin GT, et al. (October 2006). "AMG 531, a thrombopoiesis-stimulating protein, for chronic ITP". The New England Journal of Medicine. 355 (16): 1672–81. doi:10.1056/NEJMoa054626. PMID 17050891.
- ^ Kuter DJ, Rummel M, Boccia R, Macik BG, Pabinger I, Selleslag D, et al. (November 2010). "Romiplostim or standard of care in patients with immune thrombocytopenia". The New England Journal of Medicine. 363 (20): 1889–99. doi:10.1056/NEJMoa1002625. PMID 21067381. S2CID 22991485.
- ^ "AMGEN — Press Releases". Archived from the original on 2008-09-15. Retrieved 2008-08-23.
- ^ Bussel JB, Cheng G, Saleh MN, Psaila B, Kovaleva L, Meddeb B, et al. (November 2007). "Eltrombopag for the treatment of chronic idiopathic thrombocytopenic purpura". The New England Journal of Medicine. 357 (22): 2237–47. doi:10.1056/NEJMoa073275. PMID 18046028.
- ^ "FDA approves Promacta (eltrombopag), the first oral medication to increase platelet production for people with serious blood disorder" (Press release). GlaxoSmithKline. 2008-11-20. Archived from the original on 2010-12-27. Retrieved 2008-11-25.
- ^ a b c Kara-Jovanović A, Suvajdžić-Vuković N (2020-04-30). "Clinical Presentation and Therapy of Primary Immune Thrombocytopenia Resistant to Splenectomy". International Journal of Medical Students. 8 (1): 11–14. doi:10.5195/ijms.2020.436. ISSN 2076-6327.
- ^ Chaturvedi S, Arnold DM, McCrae KR (March 2018). "Splenectomy for immune thrombocytopenia: down but not out". Blood. 131 (11): 1172–82. doi:10.1182/blood-2017-09-742353. PMC 5855018. PMID 29295846.
- ^ Vianelli N, Galli M, de Vivo A, Intermesoli T, Giannini B, Mazzucconi MG, et al. (January 2005). "Efficacy and safety of splenectomy in immune thrombocytopenic purpura: long-term results of 402 cases". Haematologica. 90 (1): 72–7. PMID 15642672.
- ^ a b McMillan R, Durette C (August 2004). "Long-term outcomes in adults with chronic ITP after splenectomy failure". Blood. 104 (4): 956–60. doi:10.1182/blood-2003-11-3908. PMID 15100149.
- ^ Vianelli N, Palandri F, Polverelli N, Stasi R, Joelsson J, Johansson E, et al. (June 2013). "Splenectomy as a curative treatment for immune thrombocytopenia: a retrospective analysis of 233 patients with a minimum follow up of 10 years". Haematologica. 98 (6): 875–80. doi:10.3324/haematol.2012.075648. PMC 3669442. PMID 23144195.
- ^ Ahmed R, Devasia AJ, Viswabandya A, Lakshmi KM, Abraham A, Karl S, et al. (September 2016). "Long-term outcome following splenectomy for chronic and persistent immune thrombocytopenia (ITP) in adults and children : Splenectomy in ITP". Annals of Hematology. 95 (9): 1429–34. doi:10.1007/s00277-016-2738-3. PMID 27370992. S2CID 23025470.
- ^ Bhatt NS, Bhatt P, Donda K, Dapaah-Siakwan F, Chaudhari R, Linga VG, et al. (July 2018). "Temporal trends of splenectomy in pediatric hospitalizations with immune thrombocytopenia". Pediatric Blood & Cancer. 65 (7): e27072. doi:10.1002/pbc.27072. PMID 29637697. S2CID 4778713.
- ^ Tran, Lieu; Goel, Ruchika; Krishnamurti, Lakshmanan (2010-11-19). "Platelet Transfusions In Patients with Immune Thrombocytopenic Purpura (ITP) — Evaluation of the Current Nationwide Inpatient Hospital Practices". Blood. 116 (21): 3809. doi:10.1182/blood.V116.21.3809.3809. ISSN 0006-4971.
- ^ Stasi R, Sarpatwari A, Segal JB, Osborn J, Evangelista ML, Cooper N, et al. (February 2009). "Effects of eradication of Helicobacter pylori infection in patients with immune thrombocytopenic purpura: a systematic review". Blood. 113 (6): 1231–40. doi:10.1182/blood-2008-07-167155. PMID 18945961.
- ^ Provan D, Stasi R, Newland AC, Blanchette VS, Bolton-Maggs P, Bussel JB, et al. (January 2010). "International consensus report on the investigation and management of primary immune thrombocytopenia". Blood. 115 (2): 168–86. doi:10.1182/blood-2009-06-225565. PMID 19846889.
- ^ Godeau B, Durand JM, Roudot-Thoraval F, Tennezé A, Oksenhendler E, Kaplanski G, et al. (May 1997). "Dapsone for chronic autoimmune thrombocytopenic purpura: a report of 66 cases". British Journal of Haematology. 97 (2): 336–9. doi:10.1046/j.1365-2141.1997.412687.x. PMID 9163598. S2CID 23283382.
- ^ Wozel G, Blasum C (March 2014). "Dapsone in dermatology and beyond". Archives of Dermatological Research. 306 (2): 103–24. doi:10.1007/s00403-013-1409-7. PMC 3927068. PMID 24310318.
- ^ "Immune (idiopathic) thrombocytopenic purpura: rituximab". National Institute for Health and Care Excellence. October 2014.
- ^ "FDA approves fostamatinib tablets for ITP". FDA.gov. FDA. April 18, 2018. Retrieved 18 August 2024.
- ^ Tungjitviboonkun S, Bumrungratanayos N, Jitwimungsanon J, Kheamakulvanich T, Siramongkholkarn S (September 2024). "Efficacy and safety of fostamatinib in refractory immune thrombocytopenia: a meta-analysis from randomized controlled trials". Ann Hematol. 103 (9): 3357–68. doi:10.1007/s00277-024-05824-7. PMC 11358303. PMID 38856778.
- ^ Chen Y, Xu Y, Li H, Sun T, Cao X, Wang Y, Xue F, Liu W, Liu X, Dong H, Fu R, Dai X, Wang W, Ma Y, Song Z, Chi Y, Ju M, Gu W, Pei X, Yang R, Zhang L (June 2024). "A Novel Anti-CD38 Monoclonal Antibody for Treating Immune Thrombocytopenia". N Engl J Med. 390 (23): 2178–90. doi:10.1056/NEJMoa2400409. PMID 38899695.
- ^ "Study of Ianalumab Versus Placebo in Addition to First-line Corticosteroids in Primary Immune Thrombocytopenia (ITP) (VAYHIT1)". ClinicalTrials.gov. National Library of Medicine. Retrieved 18 August 2024.
- ^ purohit, kartik (1995). "Long-term observation of 208 adults with chronic idiopathic thrombocytopenic purpura". The American Journal of Medicine. 98 (5): 436–442. doi:10.1016/S0002-9343(99)80342-8. PMID 7733121 – via elsevier.
- ^ a b Watts RG (October 2004). "Idiopathic thrombocytopenic purpura: a 10-year natural history study at the Childrens [sic] Hospital of Alabama". Clinical Pediatrics. 43 (8): 691–702. doi:10.1177/000992280404300802. PMID 15494875. S2CID 43752571.
- ^ Treutiger I, Rajantie J, Zeller B, Henter JI, Elinder G, Rosthøj S (August 2007). "Does treatment of newly diagnosed idiopathic thrombocytopenic purpura reduce morbidity?". Archives of Disease in Childhood. 92 (8): 704–7. doi:10.1136/adc.2006.098442. PMC 2083887. PMID 17460024.
- ^ Ou CY, Hsieh KS, Chiou YH, Chang YH, Ger LP (2006). "A comparative study of initial use of intravenous immunoglobulin and prednisolone treatments in childhood idiopathic thrombocytopenic purpur". Acta Paediatrica Taiwanica = Taiwan Er Ke Yi Xue Hui Za Zhi. 47 (5): 226–31. PMID 17352309.
- ^ Nourse JP, Lea R, Crooks P, Wright G, Tran H, Catalano J, et al. (January 2012). "The KIR2DS2/DL2 genotype is associated with adult persistent/chronic and relapsed immune thrombocytopenia independently of FCGR3a-158 polymorphisms". Blood Coagulation & Fibrinolysis. 23 (1): 45–50. doi:10.1097/mbc.0b013e32834d7ce3. PMID 22024796. S2CID 3327902.
- ^ Seymour LA, Nourse JP, Crooks P, Wockner L, Bird R, Tran H, Gandhi MK (March 2014). "The presence of KIR2DS5 confers protection against adult immune thrombocytopenia". Tissue Antigens. 83 (3): 154–60. doi:10.1111/tan.12295. PMID 24571473.
- ^ Cines DB, Blanchette VS (March 2002). "Immune thrombocytopenic purpura". The New England Journal of Medicine. 346 (13): 995–1008. doi:10.1056/NEJMra010501. PMID 11919310.
- ^ "Amgen to Discuss Romiplostim BLA". drugs.com. March 12, 2008. Retrieved 2008-11-04.
- ^ Schoonen WM, Kucera G, Coalson J, Li L, Rutstein M, Mowat F, et al. (April 2009). "Epidemiology of immune thrombocytopenic purpura in the General Practice Research Database". British Journal of Haematology. 145 (2): 235–44. doi:10.1111/j.1365-2141.2009.07615.x. PMID 19245432. S2CID 45178771.
- ^ a b c d e f g h i j k l m n Bussel, James B.; Hou, Ming; Cines, Douglas B. (10 August 2023). "Management of Primary Immune Thrombocytopenia in Pregnancy". New England Journal of Medicine. 389 (6): 540–8. doi:10.1056/NEJMra2214617. PMID 37590449. S2CID 260787282.
- ^ a b Roback, John D.; Combs, Martha Rae; Grossman, Brenda J.; Hillyer, Christopher D. (2008). AABB Technical Manual (16th ed.). Bethesda: AABB Press. ISBN 978-1-56395-260-9. OCLC 1239351081.
- ^ Webert KE, Mittal R, Sigouin C, Heddle NM, Kelton JG (December 2003). "A retrospective 11-year analysis of obstetric patients with idiopathic thrombocytopenic purpura". Blood. 102 (13): 4306–11. doi:10.1182/blood-2002-10-3317. PMID 12947011. S2CID 29019817.
- ^ a b Leonard, George; Mais, Daniel D.; American Society for Clinical Pathology (2009). Quick Compendium of Clinical Pathology. American Society for Clinical Pathology Press. ISBN 978-0-89189-579-4. OCLC 489487193.
- ^ a b c d e Stasi R, Newland AC (May 2011). "ITP: a historical perspective". British Journal of Haematology. 153 (4): 437–50. doi:10.1111/j.1365-2141.2010.08562.x. PMID 21466538. S2CID 37150678.
- ^ synd/3349 at Who Named It?
- ^ Liebman HA (2008). "Immune thrombocytopenia (ITP): an historical perspective". Hematology. American Society of Hematology. Education Program. 2008 (1): 205. doi:10.1182/asheducation-2008.1.205. PMID 19074083.