
Birch reduction
| Birch reduction | |
|---|---|
| Named after | Arthur Birch |
| Reaction type | Organic redox reaction |
| Identifiers | |
| Organic Chemistry Portal | birch-reduction |
| RSC ontology ID | RXNO:0000042 |
The Birch reduction is an organic reaction that is used to convert arenes to 1,4-cyclohexadienes. The reaction is named after the Australian chemist Arthur Birch and involves the organic reduction of aromatic rings in an amine solvent (traditionally liquid ammonia) with an alkali metal (traditionally sodium) and a proton source (traditionally an alcohol). Unlike catalytic hydrogenation, Birch reduction does not reduce the aromatic ring all the way to a cyclohexane.

An example is the reduction of naphthalene in ammonia and ethanol:

Reaction mechanism and regioselectivity
[edit]A solution of sodium in liquid ammonia consists of the intensely blue electride salt [Na(NH3)x]+ e−. The solvated electrons add to the aromatic ring to give a radical anion, which then abstracts a proton from the alcohol. The process then repeats at either the ortho or para position (depending on substituents) to give the final diene.[1] The residual double bonds do not stabilize further radical additions.[2][3]

The reaction is known to be third order – first order in the aromatic, first order in the alkali metal, and first order in the alcohol.[4] This requires that the rate-limiting step be the conversion of radical anion B to the cyclohexadienyl radical C.

That step also determines the structure of the product. Although Arthur Birch originally argued that the protonation occurred at the meta position,[5] subsequent investigation has revealed that protonation occurs at either the ortho or para position. Electron donors tend to induce ortho protonation, as shown in the reduction of anisole (1). Electron-withdrawing substituents tend to induce para protonation, as shown in the reduction of benzoic acid (2).[6]


Solvated electrons will preferentially reduce sufficiently electronegative functional groups, such as ketones or nitro groups, but do not attack alcohols, carboxylic acids, or ethers.[6]
Secondary protonation regioselectivity
[edit]The second reduction and protonation also poses mechanistic questions. Thus there are three resonance structures for the carbanion (labeled B, C and D in the picture).

Simple Hückel computations lead to equal electron densities at the three atoms 1, 3 and 5, but asymmetric bond orders. Modifying the exchange integrals to account for varying interatomic distances, produces maximum electron density at the central atom 1,[7][8][9] a result confirmed by more modern RHF computations.[10]
| Approximation | Density Atom 3 | Density Atom 2 | Density Atom 1 | Bond Order 2–3 | Bond Order 1–2 |
|---|---|---|---|---|---|
| Hückel (1st approx) | 0.333 | 0.00 | 0.333 | 0.788 | 0.578 |
| 2nd approx | 0.317 | 0.00 | 0.365 | 0.802 | 0.564 |
| 3rd approx | 0.316 | 0.00 | 0.368 | 0.802 | 0.562 |
The result is analogous to conjugated enolates. When those anions (but not the enol tautomer) kinetically protonate, they do so at the center to afford the β,γ-unsaturated carbonyl.[7][11]
Modifications
[edit]Traditional Birch reduction requires cryogenic temperatures to liquify ammonia and pyrophoric alkali-metal electron donors. Variants have developed to reduce either inconvenience.
Many amines serve as alternative solvents: for example, THF[12][13] or mixed n-propylamine and ethylenediamine.[14]
To avoid direct alkali, there are chemical alternatives, such as M-SG reducing agent. The reduction can also be powered by an external potential or sacrificial anode (magnesium or aluminum), but then alkali metal salts are necessary to colocate the reactants via complexation.[15]
Birch alkylation
[edit]In Birch alkylation the anion formed in the Birch reduction is trapped by a suitable electrophile such as a haloalkane, for example:[16]

Birch Alkylation Org Synth 1990

In substituted aromatics, an electron-withdrawing substituent, such as a carboxylic acid, will stabilize the carbanion to generate the least-substituted olefin;[17] an electron-donating substituent has the opposite effect.[18]
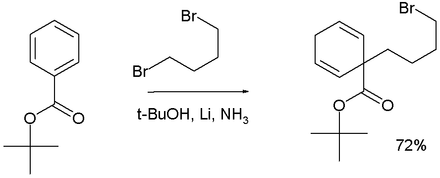
Adding 1,4-dibromobutane to a Birch reduction of tert-butyl benzoate forms the 1,1-cyclohexadiene product.[19]

{kind=link}
Benkeser reduction
[edit]The Benkeser reduction is the hydrogenation of polycyclic aromatic hydrocarbons, especially naphthalenes using lithium or calcium metal in low molecular weight alkyl amines solvents. Unlike traditional Birch reduction, the reaction can be conducted at temperatures higher than the boiling point of ammonia (−33 °C).[20][21]
For the reduction of naphthalene with lithium in a mixed ethylamine-dimethylamine solution, the principal products are bicyclo[3.3.0]dec-(1,9)-ene, bicyclo[3.3.0]dec-(1,2)-ene and bicyclo[3.3.0]decane.[22][23]
.png)

The directing effects of naphthalene substituents remain relatively unstudied theoretically. Substituents adjacent to the bridge appear to direct reduction to the unsubstituted ring; β substituents (one bond further) tend to direct reduction to the substituted ring.[6]
History
[edit]Arthur Birch, building on earlier work by Wooster and Godfrey,[24] developed the reaction while working in the Dyson Perrins Laboratory at the University of Oxford.[25] Birch's original procedure used sodium and ethanol;[5][26][27] Alfred L. Wilds later discovered that lithium gives better yields.[28][29]
The reaction was difficult to understand mechanistically, with controversy lasting into the 1990s.
The case with electron-withdrawing groups is obvious, because the Birch alkylation serves as a trap for the penultimate dianion D. This dianion appears even in alcohol-free reactions. Thus the initial protonation is para rather than ipso, as seen in the B-C transformation.[30][31][32]

For electron-donating substituents, Birch initially proposed meta attack, corresponding to the location of greatest electron density in a neutral benzene ring, a position endorsed by Krapcho and Bothner-By.[4][33] These conclusions were challenged by Zimmerman in 1961, who computed electron densities of the radical and diene anions, revealing that the ortho site which was most negative and thus most likely to protonate.[7][9] But the situation remained uncertain, because computations remained highly sensitive to transition geometry. Worse, Hückel orbital and unrestricted Hartree-Fock computations gave conflicting answers. Burnham, in 1969, concluded that the trustworthiest computations supported meta attack;[34] Birch and Radom, in 1980, concluded that both ortho and meta substitutions would occur with a slight preference for ortho.[35]
In the earlier 1990s, Zimmerman and Wang developed an experiment technique to distinguish between ortho and meta protonation. The method began with the premise that carbanions are much more basic than the corresponding radical anions and thus protonate less selectively. Correspondingly, the two protonations in Birch reduction should exhibit an isotope effect: in a protium–deuterium medium, the radical anion should preferentially protonate and the carbanion deuterate. Indeed, a variety of methoxylated aromatics exhibited less ortho deuterium than meta (a 1:7 ratio). Moreover, modern electron density computations now firmly indicated ortho protonation; frontier orbital densities, most analogous to the traditional computations used in past studies, did not.[10]
Although Birch remained reluctant to concede that ortho protonation was preferred as late as 1996,[36] Zimmerman and Wang had won the day: modern textbooks unequivocally agree that electron-donating substituents promote ortho attack.[6]
Additional reading
[edit]- Caine, D. (1976). "Reduction and Related Reactions of α,β-Unsaturated Carbonyl Compounds with Metals in Liquid Ammonia". Org. React. (review). 23: 1–258. doi:10.1002/0471264180.or023.01. ISBN 0471264180.
See also
[edit]- Solvated electron — the reducing agent
- Bouveault–Blanc reduction — another reaction using solvated electrons
- Synthesis of methamphetamine — an application
References
[edit]- ^ March, Jerry (1985), Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, 3rd edition, New York: Wiley, ISBN 9780471854722, OCLC 642506595
- ^ Rabideau, P. W.; Marcinow, Z. (1992). "The Birch Reduction of Aromatic Compounds". Org. React. (review). 42: 1–334. doi:10.1002/0471264180.or042.01. ISBN 0471264180.
- ^ Mander, L. N. (1991). "Partial Reduction of Aromatic Rings by Dissolving Metals and by Other Methods". Compr. Org. Synth. (review). 8: 489–521. doi:10.1016/B978-0-08-052349-1.00237-7. ISBN 978-0-08-052349-1.
- ^ a b Krapcho, A. P.; Bothner-By, A. A. (1959). "Kinetics of the Metal-Ammonia-Alcohol Reductions of Benzene and Substituted Benzenes1". J. Am. Chem. Soc. 81 (14): 3658–3666. doi:10.1021/ja01523a042.
- ^ a b Birch 1944.
- ^ a b c d Carey, Francis A.; Sundberg, Richard J. (2007). Advanced Organic Chemistry. Vol. B: Reactions and Synthesis (5th ed.). New York: Springer. pp. 437–439. ISBN 978-0-387-44899-2.
- ^ a b c Zimmerman, H. E. (1961). "Orientation in Metal Ammonia Reductions". Tetrahedron. 16 (1–4): 169–176. doi:10.1016/0040-4020(61)80067-7.
- ^ Zimmerman, Howard E (1975). Quantum Mechanics for Organic Chemists. New York: Academic Press. pp. 154–5. ISBN 0-12-781650-X.
- ^ a b Zimmerman, H. E. (1963). "Base-Catalyzed Rearrangements". In De Mayo, P. (ed.). Molecular Rearrangements. New York: Interscience. pp. 350–352.
- ^ a b
- Zimmerman, H. E.; Wang, P. A. (1990). "The Regioselectivity of the Birch Reduction". J. Am. Chem. Soc. 112 (3): 1280–1281. doi:10.1021/ja00159a078.
- Zimmerman, H. E.; Wang, P. A. (1993). "Regioselectivity of the Birch Reduction". J. Am. Chem. Soc. 115 (6): 2205–2216. doi:10.1021/ja00059a015.
- ^ Paufler, R. M. (1960) Ph.D. Thesis, Northwestern University, Evanston, IL.
- ^ Ecsery, Zoltan & Muller, Miklos (1961). "Reduction vitamin D2 with alkaly metals". Magyar Kémiai Folyóirat. 67: 330–332.
- ^ Donohoe, Timothy J. & House, David (2002). "Ammonia Free Partial Reduction of Aromatic Compounds Using Lithium Di-tert-butylbiphenyl (LiDBB)". Journal of Organic Chemistry. 67 (14): 5015–5018. doi:10.1021/jo0257593. PMID 12098328.
- ^ Garst, Michael E.; Lloyd J.; Shervin; N. Andrew; Natalie C.; Alfred A.; et al. (2000). "Reductions with Lithium in Low Molecular Weight Amines and Ethylenediamine". Journal of Organic Chemistry. 65 (21): 7098–7104. doi:10.1021/jo0008136. PMID 11031034.
- ^ Peters, Byron K.; Rodriguez, Kevin X.; Reisberg, Solomon H.; Beil, Sebastian B.; Hickey, David P.; Kawamata, Yu; Collins, Michael; Starr, Jeremy; Chen, Longrui; Udyavara, Sagar; Klunder, Kevin; Gorey, Timothy J.; Anderson, Scott L.; Neurock, Matthew; Minteer, Shelley D.; Baran, Phil S. (21 February 2019). "Scalable and safe synthetic organic electroreduction inspired by Li-ion battery chemistry". Science. 363 (6429): 838–845. Bibcode:2019Sci...363..838P. doi:10.1126/science.aav5606. PMC 7001862. PMID 30792297.
- ^ Taber, D. F.; Gunn, B. P.; Ching Chiu, I. (1983). "Alkylation of the anion from Birch reduction of o-Anisic acid: 2-Heptyl-2-cyclohexenone". Organic Syntheses; Collected Volumes, vol. 7, p. 249.
- ^ Kuehne, M. E.; Lambert, B. F. (1963). "1,4-Dihydrobenzoic acid". Organic Syntheses; Collected Volumes, vol. 5, p. 400.
- ^ Paquette, L. A.; Barrett, J. H. (1969). "2,7-Dimethyloxepin". Organic Syntheses; Collected Volumes, vol. 5, p. 467.
- ^ Clive, Derrick L. J. & Sunasee, Rajesh (2007). "Formation of Benzo-Fused Carbocycles by Formal Radical Cyclization onto an Aromatic Ring". Organic Letters. 9 (14): 2677–2680. doi:10.1021/ol070849l. PMID 17559217.
- ^ Birch Reductions, Institute of Chemistry, Skopje, Macedonia
- ^ Vogel, E.; Klug, W.; Breuer, A. (1974). "1,6-Methano[10]annulene". Organic Syntheses; Collected Volumes, vol. 6.
- ^ Edwin M. Kaiser and Robert A. Benkeser "Δ9,10-Octalin" Org. Synth. 1970, vol. 50, p. 88ff. doi:10.15227/orgsyn.050.0088
- ^ Merck Index, 13th Ed.
- ^ Wooster, C. B.; Godfrey, K. L. (1937). "Mechanism of the Reduction of Unsaturated Compounds with Alkali Metals and Water". Journal of the American Chemical Society. 59 (3): 596. doi:10.1021/ja01282a504.
- ^
- Birch, A. J. (1944). "Reduction by dissolving metals. Part I". J. Chem. Soc.: 430. doi:10.1039/JR9440000430.
- Birch, A. J. (1945). "Reduction by dissolving metals. Part II". J. Chem. Soc.: 809. doi:10.1039/jr9450000809.
- Birch, A. J. (1946). "Reduction by dissolving metals. Part III". J. Chem. Soc.: 593. doi:10.1039/jr9460000593.
- Birch, A. J. (1947). "Reduction by dissolving metals. Part IV". J. Chem. Soc.: 102. doi:10.1039/jr9470000102.
- Birch, Arthur J. (1947). "Reduction by dissolving metals. Part V". J. Chem. Soc.: 1642. doi:10.1039/jr9470001642.
- Birch, A. J.; Mukherji, S. M. (1949). "Reduction by dissolving metals. Part VI. Some applications in synthesis". J. Chem. Soc.: 2531. doi:10.1039/jr9490002531.
- ^ Birch 1945.
- ^ Birch 1946.
- ^ Wilds, A. L.; Nelson, N. A. (1953). "A Superior Method for Reducing Phenol Ethers to Dihydro Derivatives and Unsaturated Ketones". J. Am. Chem. Soc. 75 (21): 5360–5365. doi:10.1021/ja01117a064.
- ^ Birch, A. J.; Smith, H. (1958). "Reduction by metal–amine solutions: applications in synthesis and determination of structure". Quart. Rev. (review). 12 (1): 17. doi:10.1039/qr9581200017.
- ^ Bachi, J. W.; Epstein, Y.; Herzberg-Minzly, H.; Loewnenthal, J. E. (1969). "Synthesis of compounds related to gibberellic acid. III. Analogs of ring a of the gibberellins". J. Org. Chem. 34: 126–135. doi:10.1021/jo00838a030.
- ^ Taber, D. F.; Gunn, B.P; Ching Chiu, I (1983). "Alkylation of the Anion from Birch Reduction of o-Anisic Acid: 2-Heptyl-2-Cyclohexenone". Organic Syntheses. 61: 59; Collected Volumes, vol. 7, p. 249.
- ^ Guo, Z.; Schultz, A. G. (2001). "Organic synthesis methodology. Preparation and diastereoselective birch reduction-alkylation of 3-substituted 2-methyl-2,3-dihydroisoindol-1-ones". J. Org. Chem. 66 (6): 2154–2157. doi:10.1021/jo005693g. PMID 11300915.
- ^ Birch, A. J.; Nasipuri, D. (1959). "Reaction mechanisms in reduction by metal-ammonia solutions". Tetrahedron. 6 (2): 148–153. doi:10.1016/0040-4020(59)85008-0.
- ^ Burnham, D. R. (1969). "Orientation in the mechanism of the Birch reduction of anisole". Tetrahedron. 25 (4): 897–904. doi:10.1016/0040-4020(69)85023-4.
- ^
- Birch, A. J.; Hinde, A. L.; Radom, L. (1980). "A theoretical approach to the Birch reduction. Structures and stabilities of the radical anions of substituted benzenes". J. Am. Chem. Soc. 102 (10): 3370–3376. doi:10.1021/ja00530a012.
- Birch, A. J.; Radom, L. (1980). "A theoretical approach to the Birch reduction. Structures and stabilities of cyclohexadienyl radicals". J. Am. Chem. Soc. 102 (12): 4074–4080. doi:10.1021/ja00532a016.
- ^ See diagrams in:
- Birch, A. J. (1992). "Steroid hormones and the Luftwaffe. A venture into fundamental strategic research and some of its consequences: The Birch reduction becomes a birth reduction". Steroids. 57 (8): 363–377. doi:10.1016/0039-128X(92)90080-S. PMID 1519267. S2CID 24827957.
- Birch, A. J. (1996). "The Birch reduction in organic synthesis". Pure Appl. Chem. 68 (3): 553–556. doi:10.1351/pac199668030553. S2CID 41494178.
| Authority control databases: National |
|---|