
Spinocerebellar ataxia
| Spinocerebellar ataxia | |
|---|---|
| Other names | Spinocerebellar atrophy or Spinocerebellar degeneration |
 | |
| Cerebellum (in blue) of the human brain | |
| Specialty | Neurology |
Spinocerebellar ataxia (SCA) is a progressive, degenerative,[1] genetic disease with multiple types, each of which could be considered a neurological condition in its own right. An estimated 150,000 people in the United States have a diagnosis of spinocerebellar ataxia at any given time. SCA is hereditary, progressive, degenerative, and often fatal. There is no known effective treatment or cure. SCA can affect anyone of any age. The disease is caused by either a recessive or dominant gene. In many cases people are not aware that they carry a relevant gene until they have children who begin to show signs of having the disorder.[2] Currently, research is being conducted at Universities, such as the University of Minnesota, to elucidate many of the unknown characteristics of the disease.[3]
Signs and symptoms
[edit]Spinocerebellar ataxia (SCA) is one of a group of genetic disorders characterized by slowly progressive incoordination of gait and is often associated with poor coordination of hands, speech, and eye movements. A review of different clinical features among SCA subtypes was recently published describing the frequency of non-cerebellar features, like parkinsonism, chorea, pyramidalism, cognitive impairment, peripheral neuropathy, seizures, among others.[4] As with other forms of ataxia, SCA frequently results in atrophy of the cerebellum,[5] loss of fine coordination of muscle movements leading to unsteady and clumsy motion, and other symptoms. Ocular deficits can be quantified using the SODA scale.[6]
The symptoms of an ataxia vary with the specific type and with the individual patient. Many subtypes of spinocerebellar ataxia result in cases where an individual retains full mental capacity but progressively loses physical control, but nearly half of the identified subtypes result in cognitive dysfunction, dementia, and mental retardation.[7]
Cause
[edit]The hereditary ataxias are categorized by mode of inheritance and causative gene or chromosomal locus. The hereditary ataxias can be inherited in an autosomal dominant, autosomal recessive, or X-linked manner.[citation needed]
- Many types of autosomal dominant cerebellar ataxias for which specific genetic information is available are now known. Synonyms for autosomal-dominant cerebellar ataxias (ADCA) used prior to the current understanding of the molecular genetics were Marie's ataxia, inherited olivopontocerebellar atrophy, cerebello-olivary atrophy, or the more generic term "spinocerebellar degeneration." (Spinocerebellar degeneration is a rare inherited neurological disorder of the central nervous system characterized by the slow degeneration of certain areas of the brain. There are three forms of spinocerebellar degeneration: Types 1, 2, 3. Symptoms begin during adulthood.)[citation needed]
- There are five typical autosomal-recessive disorders in which ataxia is a prominent feature: Friedreich ataxia, ataxia-telangiectasia, ataxia with vitamin E deficiency, ataxia with oculomotor apraxia (AOA), spastic ataxia. Disorder subdivisions: Friedreich's ataxia, spinocerebellar ataxia, ataxia telangiectasia, vasomotor ataxia, vestibulocerebellar, ataxiadynamia, ataxiophemia, and olivopontocerebellar atrophy.[citation needed]
- There have been reported cases where a polyglutamine expansion may lengthen when passed down, which often can result in an earlier age-of-onset and a more severe disease phenotype for individuals who inherit the disease allele. This falls under the category of genetic anticipation.[8][9] Several types of SCA are characterized by repeat expansion of the trinucleotide sequence CAG in DNA that encodes a polyglutamine repeat tract in protein. The expansion of CAG repeats over successive generations appears to be due to slipped strand mispairing during DNA replication or DNA repair.[10]
-
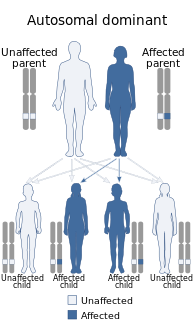 There are numerous types of autosomal-dominant cerebellar ataxias
There are numerous types of autosomal-dominant cerebellar ataxias -
 There are five typical autosomal recessive disorders in which ataxia is a prominent feature
There are five typical autosomal recessive disorders in which ataxia is a prominent feature


Diagnosis
[edit]Classification
[edit]A few SCAs remain unspecified and can not be precisely diagnosed, but in the last decade[as of?] genetic testing has allowed precise identification of dozens of different SCAs and more tests are being added each year.[11] In 2008, a genetic ataxia blood test developed to test for 12 types of SCA, Friedreich's ataxia, and several others. However, since not every SCA has been genetically identified some SCAs are still diagnosed by neurological examination, which may include a physical exam, family history, MRI scanning of the brain and spine, and spinal tap.[12]
Many SCAs below fall under the category of polyglutamine diseases, which are caused when a disease-associated protein (i.e., ataxin-1, ataxin-3, etc.) contains a large number of repeats of glutamine residues, termed a polyQ sequence or a "CAG trinucleotide repeat" disease for either the one-letter designation or codon for glutamine respectively. The threshold for symptoms in most forms of SCA is around 35, though for SCA3 it extends beyond 50. Most polyglutamine diseases are dominant due to the interactions of resulting polyQ tail.[citation needed]
The first ataxia gene was identified in 1993 and called "Spinocerebellar ataxia type 1" (SCA1); later genes were called SCA2, SCA3, etc. Usually, the "type" number of "SCA" refers to the order in which the gene was found. At this time, there are at least 49 different gene mutations that have been found.[citation needed]
The following is a list of some of the many types of Spinocerebellar ataxia.
| SCA Type | Average Onset (Range in Years) |
Average Duration (Range in Years) |
What the patient experiences | Common origin | Problems with DNA |
|---|---|---|---|---|---|
| SCA1[13] (ATXN1) | 4th decade (<10 to >60) |
15 years (10–35) |
Hypermetric saccades, slow saccades, upper motor neuron (note: saccades relates to eye movement) |
CAG repeat, 6p (Ataxin 1) | |
| SCA2[14] (ATXN2) | 3rd–4th decade (<10 to >60) |
10 years (1–30) |
Diminished velocity saccades areflexia (absence of neurologic reflexes) |
Cuba | CAG repeat, 12q |
| SCA3[15] (MJD) (ATXN3) | 4th decade (10–70) |
10 years (1–20) |
Also called Machado-Joseph disease (MJD)[16] Gaze-evoked nystagmus (a rapid, involuntary, oscillatory motion of the eyeball) upper motor neuron slow saccades |
Azores (Portugal) |
CAG repeat, 14q |
| SCA4 (PLEKHG4) | 4th–7th decade (19–72) |
Decades | areflexia (absence of neurologic reflexes) | Chromosome 16q | |
| SCA5 (SPTBN2) | 3rd–4th decade (10–68) |
>25 years | Pure cerebellar | Chromosome 11 | |
| SCA6[17] (CACNA1A) | 5th–6th decade (19–71) |
>25 years | Downbeating nystagmus, positional vertigo Symptoms can appear for the first time as late as 65 years old. |
CAG repeat, 19p Calcium channel gene | |
| SCA7[18] (ATXN7) | 3rd–4th decade (0.5–60) |
20 years (1–45; early onset correlates with shorter duration) |
Macular degeneration, upper motor neuron, slow saccades | CAG repeat, 3p (Ataxin 7) | |
| SCA8[19] (IOSCA) | 39 yrs (18–65) |
Normal lifespan | Horizontal nystagmus (a rapid, involuntary, oscillatory motion of the eyeball), instability, lack of coordination | CTG repeat,[20] 13q | |
| SCA10[21] (ATXN10) | 36 years | 9 years | ataxia, seizures | Mexico | Chromosome 22q linked pentanucleotide repeat |
| SCA11 (TTBK2) | 30 yrs (15–70) |
Normal lifespan | Mild, remain ambulatory (able to walk about on one's own) | 15q | |
| SCA12[22] (PPP2R2B) | 33 yrs (8–55) |
Head and hand tremor, akinesia (loss of normal motor function, resulting in impaired muscle movement) |
CAG repeat, 5q | ||
| SCA13 (KCNC3) | Childhood or adulthood depending on mutation | Depending on KCNC3 (a kind of gene) | Intellectual disability | 19q | |
| SCA14[23] (PRKCG) | 28 yrs (12–42) |
Decades (1–30) |
Myoclonus (a sudden twitching of muscles or parts of muscles, without any rhythm or pattern, occurring in various brain disorders) | 19q | |
| SCA16 (ITPR1) | 39 yrs (20–66) |
1–40 years | Head and hand tremor | 8q | |
| SCA17 (TBP) | CAG repeat, 6q (TATA-binding protein) | ||||
| SCA19, SCA22 (KCND3[24]) | Mild cerebellar syndrome, dysarthria | ||||
| SCA25 | 1.5–39 yrs | Unknown | ataxia with sensory neuropathy, vomiting and gastrointestinal pain. | 2p | |
| SCA27[25] (FGF14[24]) | 15–20 yrs | Unknown | ataxia with poor cognition, dyskinesias and tremor. | FGF14 13q34 | |
| SCA35 | 40–48 years | Unknown | gait and limb ataxia, dysarthria, ocular dysmetria, intention tremor, pseudobulbar palsy, spasmodic torticollis, extensor plantar responses, reduced proprioception and hyperreflexia | China | transglutaminase 6 (TGM6) located at chromosome 20p13 |
| SCA36 | 5th and 6th decade (adulthood) | Decades | ataxia, hyperrheflexia, dysarthria, fasciculations of the tongue with subsequent wasting of the tongue | NOP56 | |
| SCA37 | Adulthood | Decades | dysarthria, slowly progressive gait and limb ataxia with severe dysmetria in the lower extremities, mild dysmetria in the upper extremities, dysphagia, and abnormal ocular movements | DAB1 |
Others include SCA18, SCA20, SCA21, SCA23, SCA26, SCA28, and SCA29.
Four X-linked types have been described ( 302500, 302600, 301790, 301840), but only the first of these has so far been tied to a gene (SCAX1).
| Name | OMIM | RareDiseases | Other |
|---|---|---|---|
| Anemia, sideroblastic spinocerebellar ataxia; Pagon Bird Detter syndrome | 301310 | Disease ID 668 at NIH's Office of Rare Diseases | |
| Friedreich's ataxia; Spinocerebellar ataxia, Friedreich | 229300 | Disease ID 6468 at NIH's Office of Rare Diseases | |
| Infantile onset Spinocerebellar ataxia | 605361 | Disease ID 4062 at NIH's Office of Rare Diseases | |
| Spinocerebellar ataxia 1 | 164400 | Disease ID 4071 at NIH's Office of Rare Diseases | |
| Spinocerebellar ataxia 2 | 183090 | Disease ID 4072 at NIH's Office of Rare Diseases | |
| Spinocerebellar ataxia 3; Machado Joseph disease | 109150 | Disease ID 6801 at NIH's Office of Rare Diseases | |
| Spinocerebellar ataxia 4 | 600223 | Disease ID 9970 at NIH's Office of Rare Diseases | |
| Spinocerebellar ataxia 5 | 600224 | Disease ID 4953 at NIH's Office of Rare Diseases | |
| Spinocerebellar ataxia 7 | 164500 | Disease ID 4955 at NIH's Office of Rare Diseases | |
| Spinocerebellar ataxia 8 | 603680 | Disease ID 4956 at NIH's Office of Rare Diseases | |
| Spinocerebellar ataxia 13 | 605259 | Disease ID 9611 at NIH's Office of Rare Diseases | |
| Spinocerebellar ataxia 18 | 607458 | Disease ID 9976 at NIH's Office of Rare Diseases | |
| Spinocerebellar ataxia 19 | 607346 | Disease ID 9969 at NIH's Office of Rare Diseases | |
| Spinocerebellar ataxia 20 | 608687 | Disease ID 9997 at NIH's Office of Rare Diseases | |
| Spinocerebellar ataxia 21 | 607454 | Disease ID 9999 at NIH's Office of Rare Diseases | |
| Spinocerebellar ataxia 23 | 610245 | Disease ID 9950 at NIH's Office of Rare Diseases | |
| Spinocerebellar ataxia 25 | 608703 | Disease ID 9996 at NIH's Office of Rare Diseases | |
| Spinocerebellar ataxia 26 | 609306 | Disease ID 9995 at NIH's Office of Rare Diseases | |
| Spinocerebellar ataxia 28 | 610246 | Disease ID 9951 at NIH's Office of Rare Diseases | |
| Spinocerebellar ataxia 30 | 117360 | Disease ID 9975 at NIH's Office of Rare Diseases | |
| Spinocerebellar ataxia 35 | 613908 | Disease ID at NIH's Office of Rare Diseases | |
| Spinocerebellar ataxia amyotrophy deafness syndrome | Disease ID 2451 at NIH's Office of Rare Diseases | ORPHA:2074 at Orphanet | |
| Spinocerebellar ataxia, autosomal recessive 1 | 606002 | Disease ID 4949 at NIH's Office of Rare Diseases | |
| Spinocerebellar ataxia, autosomal recessive 3 | 271250 | Disease ID 9971 at NIH's Office of Rare Diseases | |
| Spinocerebellar ataxia, autosomal recessive 4 | 607317 | Disease ID 4952 at NIH's Office of Rare Diseases | |
| Spinocerebellar ataxia, autosomal recessive 5 | 606937 | Disease ID 9977 at NIH's Office of Rare Diseases | |
| Spinocerebellar ataxia, autosomal recessive 6 | 608029 | Disease ID 4954 at NIH's Office of Rare Diseases | |
| Spinocerebellar ataxia, autosomal recessive 21 - mutation in SCYL1 | Online Mendelian Inheritance in Man (OMIM): 616719 | ORPHA:466794 | |
| Spinocerebellar ataxia, autosomal recessive, with axonal neuropathy | 607250 | Disease ID 10000 at NIH's Office of Rare Diseases | |
| Spinocerebellar ataxia, X-linked, 2 | 302600 | Disease ID 9978 at NIH's Office of Rare Diseases | |
| Spinocerebellar ataxia, X-linked, 3 | 301790 | Disease ID 9981 at NIH's Office of Rare Diseases | |
| Spinocerebellar ataxia, X-linked, 4 | 301840 | Disease ID 9980 at NIH's Office of Rare Diseases |
Treatment
[edit]Medication
[edit]There is no cure for spinocerebellar ataxia, which is currently considered to be a progressive and irreversible disease, although not all types cause equally severe disability.[26]
In general, treatments are directed towards alleviating symptoms, not the disease itself. Many patients with hereditary or idiopathic forms of ataxia have other symptoms in addition to ataxia. Medications or other therapies might be appropriate for some of these symptoms, which could include tremor, stiffness, depression, spasticity, and sleep disorders, among others. Both onset of initial symptoms and duration of disease are variable. If the disease is caused by a polyglutamine trinucleotide repeat CAG expansion, a longer expansion may lead to an earlier onset and a more radical progression of clinical symptoms. Typically, a person with this disease will eventually be unable to perform daily tasks (ADLs).[27] However, rehabilitation therapists can help patients to maximize their ability of self-care and delay deterioration to certain extent.[28] Researchers are exploring multiple avenues for a cure including RNA interference (RNAi) technology, the use of stem cells, and several other avenues.[29]
On January 18, 2017, BioBlast Pharma announced completion of Phase 2a clinical trials of their medication, trehalose, in the treatment of SCA3. BioBlast has received FDA Fast Track status and orphan drug status for their treatment. The information provided by BioBlast in their research indicates that they hope this treatment may prove efficacious in other SCA treatments that have similar pathology related to PolyA and PolyQ diseases.[30][31]
In addition, Dr. Beverly Davidson has been working on a methodology using RNAi technology to find a potential cure for over 2 decades.[32] Her research began in the mid-1990s and progressed to work with mouse models about a decade later and most recently has moved to a study with non-human primates. The results from her most recent research "are supportive of clinical application of this gene therapy".[33]
Finally, another gene transfer technology discovered in 2011 has also been shown by Boudreau et al. to hold great promise and offers yet another avenue to a potential future cure.[34]
N-Acetyl-Leucine
[edit]N-Acetyl-Leucine is an orally administered, modified amino acid that is being developed as a novel treatment for multiple rare and common neurological disorders by IntraBio Inc (Oxford, United Kingdom).[35]
N-Acetyl-Leucine has been granted multiple orphan drug designations from the U.S. Food & Drug Administration (FDA)[36] and the European Medicines Agency (EMA)[37] for the treatment of various genetic diseases, including spinocerebellar ataxias. N-Acetyl-Leucine has also been granted Orphan Drug Designations in the US and EU for the related inherited cerebellar ataxia ataxia-telangiectasia U.S. Food & Drug Administration (FDA)[38] and the European Medicines Agency (EMA).[39]
Published case series studies have demonstrated the effects of acute treatment with N-Acetyl-Leucine for the treatment of inherited cerebellar ataxias, including spinocerebellar ataxias.[40][41] These studies further demonstrated that the treatment is well tolerated, with a good safety profile.[citation needed] A multinational clinical trial investigating N-Acetyl-L-Leucine for the treatment of a related inherited cerebellar ataxia, ataxia-telangiectasia, began in 2019.[42]
IntraBio is also conducting parallel clinical trials with N-Acetyl-L-Leucine for the treatment of Niemann-Pick disease type C[43] and GM2 gangliosidosis (Tay-Sachs and Sandhoff disease).[44] Future opportunities to develop N-Acetyl-Leucine include Lewy body dementia,[45] amyotrophic lateral sclerosis, restless leg syndrome, multiple sclerosis, and migraine.[46]
Rehabilitation
[edit]Physical therapists can assist patients in maintaining their level of independence through therapeutic exercise programmes. One recent research report demonstrated a gain of two SARA points (Scale for the Assessment and Rating of Ataxia) from physical therapy.[47] In general, physical therapy emphasises postural balance and gait training for ataxia patients.[48] General conditioning such as range-of-motion exercises and muscle strengthening would also be included in therapeutic exercise programmes. Research showed that spinocerebellar ataxia 2 (SCA2) patients[49] with a mild stage of the disease gained significant improvement in static balance and neurological indices after six months of a physical therapy exercise training program.[50] Occupational therapists may assist patients with incoordination or ataxia issues through the use of adaptive devices. Such devices may include a cane, crutches, walker, or wheelchair for those with impaired gait. Other devices are available to assist with writing, feeding, and self care if hand and arm coordination are impaired. A randomised clinical trial revealed that an intensive rehabilitation program with physical and occupational therapies for patients with degenerative cerebellar diseases can significantly improve functional gains in ataxia, gait, and activities of daily living. Some level of improvement was shown to be maintained 24 weeks post-treatment.[51] Speech language pathologists may use both behavioral intervention strategies as well as augmentative and alternative communication devices to help patients with impaired speech.[citation needed]
References
[edit]- ^ "spinocerebellar ataxia" at Dorland's Medical Dictionary
- ^ "Ataxias and Cerebellar or Spinocerebellar Degeneration Information Page". National Institute on Neurological Disorders and Stroke.
- ^ "Spinocerebellar Ataxia | Research Computing". rc.umn.edu. Retrieved 2024-09-17.
- ^ Rossi M, Perez-Lloret S, Doldan L, Cerquetti D, Balej J, Millar Vernetti P, et al. (April 2014). "Autosomal dominant cerebellar ataxias: a systematic review of clinical features". European Journal of Neurology. 21 (4): 607–615. doi:10.1111/ene.12350. hdl:11336/30194. PMID 24765663. S2CID 74661673.
- ^ "Spinocerebellar ataxia". Genes and Disease [Internet]. Bethesda MD: National Center for Biotechnology Information. 1998. NBK22234. — Gives a concise description of SCA, along with a picture of shrunken degenerated cerebellum.
- ^ Shaikh AG, Kim JS, Froment C, Koo YJ, Dupre N, Hadjivassiliou M, et al. (December 2022). "Scale for Ocular motor Disorders in Ataxia (SODA)". Journal of the Neurological Sciences. 443: 120472. doi:10.1016/j.jns.2022.120472. PMID 36403298. S2CID 253156325.
- ^ Teive HA, Arruda WO (2009). "Cognitive dysfunction in spinocerebellar ataxias". Dementia & Neuropsychologia. 3 (3): 180–187. doi:10.1590/S1980-57642009DN30300002. PMC 5618971. PMID 29213626.
- ^ Khristich AN, Mirkin SM (March 2020). "On the wrong DNA track: Molecular mechanisms of repeat-mediated genome instability". The Journal of Biological Chemistry. 295 (13): 4134–4170. doi:10.1074/jbc.REV119.007678. PMC 7105313. PMID 32060097.
- ^ Figueroa KP, Gross C, Buena-Atienza E, Paul S, Gandelman M, Kakar N, et al. (June 2024). "A GGC-repeat expansion in ZFHX3 encoding polyglycine causes spinocerebellar ataxia type 4 and impairs autophagy". Nature Genetics. 56 (6): 1080–1089. doi:10.1038/s41588-024-01719-5. PMID 38684900.
- ^ Usdin K, House NC, Freudenreich CH (2015). "Repeat instability during DNA repair: Insights from model systems". Critical Reviews in Biochemistry and Molecular Biology. 50 (2): 142–167. doi:10.3109/10409238.2014.999192. PMC 4454471. PMID 25608779.
- ^ "FREQUENTLY ASKED QUESTIONS ABOUT... Gene Testing for Hereditary Ataxia" (PDF). Archived from the original (PDF) on 2015-07-27. Retrieved 2017-01-25.
- ^ www.ataxia.org[full citation needed]
- ^ sca1 at NIH/UW GeneTests
- ^ sca2 at NIH/UW GeneTests
- ^ sca3 at NIH/UW GeneTests
- ^ Spinocerebellar Ataxias including Machado-Joseph Disease at NINDS
- ^ sca6 at NIH/UW GeneTests
- ^ sca7 at NIH/UW GeneTests
- ^ sca8 at NIH/UW GeneTests
- ^ Mosemiller AK, Dalton JC, Day JW, Ranum LP (2003). "Molecular genetics of spinocerebellar ataxia type 8 (SCA8)". Cytogenetic and Genome Research. 100 (1–4): 175–183. doi:10.1159/000072852. PMID 14526178. S2CID 2292926.
- ^ sca10 at NIH/UW GeneTests
- ^ sca12 at NIH/UW GeneTests
- ^ sca14 at NIH/UW GeneTests
- ^ a b Perlman SL (2016). Evaluation and Management of Ataxic Disorders: An Overview for Physicians. Minneapolis: National Ataxia Foundation. p. 6. ISBN 978-0-943218-14-4. LCCN 2007923539.
- ^ Online Mendelian Inheritance in Man (OMIM): 609307
- ^ Jiang B, Glover JN, Weinfeld M (January 2017). "Neurological disorders associated with DNA strand-break processing enzymes". Mechanisms of Ageing and Development. 161 (Pt A): 130–140. doi:10.1016/j.mad.2016.07.009. PMC 5266678. PMID 27470939.
- ^ Cruts M, Engelborghs S, van der Zee J, Van Broeckhoven C (1993). "C9orf72-Related Amyotrophic Lateral Sclerosis and Frontotemporal Dementia". In Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJ, Stephens K, Amemiya A (eds.). GeneReviews. University of Washington, Seattle. PMID 25577942.
- ^ Synofzik M, Ilg W (2014). "Motor training in degenerative spinocerebellar disease: ataxia-specific improvements by intensive physiotherapy and exergames". BioMed Research International. 2014: 583507. doi:10.1155/2014/583507. PMC 4022207. PMID 24877117.
- ^ Wilmot G. "Update on SCA Research". National Ataxia Foundation. Archived from the original on 2016-11-19. Retrieved 2017-01-26.
- ^ "Bioblast Announces Phase 2a Results of Trehalose in Patients with Spinocerebellar Ataxia Type 3 (SCA3)". Investors Hub. Retrieved 14 October 2017.
- ^ "The Orphan Genetic Disease Company: Bioblast Pharma Ltd. June 2016" (PDF). Bioblast Pharma Ltd. Retrieved 14 October 2017.
- ^ Veritas G (17 August 2013). "RNA Interference for Treating Huntington's Disease: An Interview with Dr. Beverly Davidson". Vimeo. Retrieved 14 October 2017.
- ^ Keiser MS, Kordower JH, Gonzalez-Alegre P, Davidson BL (December 2015). "Broad distribution of ataxin 1 silencing in rhesus cerebella for spinocerebellar ataxia type 1 therapy". Brain. 138 (Pt 12): 3555–3566. doi:10.1093/brain/awv292. PMC 4840549. PMID 26490326.
- ^ Boudreau RL, Spengler RM, Davidson BL (December 2011). "Rational design of therapeutic siRNAs: minimizing off-targeting potential to improve the safety of RNAi therapy for Huntington's disease". Molecular Therapy. 19 (12): 2169–2177. doi:10.1038/mt.2011.185. PMC 3242660. PMID 21952166.
- ^ "IntraBio". Archived from the original on 2019-08-01. Retrieved 2019-08-01.
- ^ "Search Orphan Drug Designations and Approvals". www.accessdata.fda.gov. Retrieved 2019-08-01.
- ^ Francisco EM (2018-12-20). "EU/3/18/2059". European Medicines Agency. Retrieved 2019-08-01.
- ^ "Search Orphan Drug Designations and Approvals". www.accessdata.fda.gov. Retrieved 2019-08-01.
- ^ "Search Orphan Drug Designations and Approvals". www.accessdata.fda.gov. Retrieved 2019-08-01.
- ^ Cross J (April 2006). "MEDLINE, PubMed, PubMed Central, and the NLM". Editors' Bulletin. 2 (1): 1–5. doi:10.1080/17521740701702115.
- ^ Schniepp R, Strupp M, Wuehr M, Jahn K, Dieterich M, Brandt T, et al. (December 2016). "Acetyl-DL-leucine improves gait variability in patients with cerebellar ataxia-a case series". Cerebellum & Ataxias. 3 (1): 8. doi:10.1186/s40673-016-0046-2. PMC 4828858. PMID 27073690.
- ^ Clinical trial number NCT03759678 for "N-Acetyl-L-Leucine for Ataxia-Telangiectasia (A-T)" at ClinicalTrials.gov
- ^ Clinical trial number NCT03759639 for "N-Acetyl-L-Leucine for Niemann-Pick Disease, Type C (NPC)" at ClinicalTrials.gov
- ^ Clinical trial number NCT03759665 for "N-Acetyl-L-Leucine for GM2 Gangliosdisosis (Tay-Sachs and Sandhoff Disease)" at ClinicalTrials.gov
- ^ "IntraBio". Archived from the original on 2019-08-01. Retrieved 2019-08-01.
- ^ Strupp M, Bayer O, Feil K, Straube A (February 2019). "Prophylactic treatment of migraine with and without aura with acetyl-DL-leucine: a case series". Journal of Neurology. 266 (2): 525–529. doi:10.1007/s00415-018-9155-6. PMID 30547273. S2CID 56148131.
- ^ Synofzik M, Ilg W (2014). "Motor training in degenerative spinocerebellar disease: ataxia-specific improvements by intensive physiotherapy and exergames". BioMed Research International. 2014: 583507. doi:10.1155/2014/583507. PMC 4022207. PMID 24877117.
- ^ Marsden J, Harris C (March 2011). "Cerebellar ataxia: pathophysiology and rehabilitation". Clinical Rehabilitation. 25 (3): 195–216. doi:10.1177/0269215510382495. PMID 21321055. S2CID 40374830.
- ^ "SCA2 information sheet from www.ataxia.org" (PDF). Archived from the original (PDF) on 2012-07-12. Retrieved 2012-05-10.
- ^ Trujillo-Martín MM, Serrano-Aguilar P, Monton-Alvarez F, Carrillo-Fumero R (June 2009). "Effectiveness and safety of treatments for degenerative ataxias: a systematic review". Movement Disorders. 24 (8): 1111–1124. doi:10.1002/mds.22564. PMID 19412936. S2CID 11008654.
- ^ Miyai I, Ito M, Hattori N, Mihara M, Hatakenaka M, Yagura H, et al. (June 2012). "Cerebellar ataxia rehabilitation trial in degenerative cerebellar diseases". Neurorehabilitation and Neural Repair. 26 (5): 515–522. doi:10.1177/1545968311425918. PMID 22140200. S2CID 23764699.
Further reading
[edit]- Perlman S (23 January 2014). "Hereditary Ataxia Overview". In Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJ, Stephens K, Amemiya A (eds.). GeneReviews [Internet]. Seattle WA: University of Washington, Seattle. PMID 20301317. NBK1138.
- Moreira MC, Koenig M (December 8, 2011). "Ataxia with Oculomotor Apraxia Type 2". In Adam MP, Feldman J, Mirzaa GM, et al. (eds.). GeneReviews [Internet]. University of Washington, Seattle. PMID 20301333. NBK1154.
- Pulst SM (1 March 2012). "Spinocerebellar Ataxia Type 13". GeneReviews [Internet]. Seattle WA: University of Washington, Seattle. PMID 20301404. NBK1225.
- Brussino A, Brusco A, Dürr A (7 February 2013). "Spinocerebellar Ataxia Type 28". In Adam MP, Feldman J, Mirzaa GM, et al. (eds.). GeneReviews [Internet]. Seattle WA: University of Washington, Seattle. PMID 21595125. NBK54582.
- Nikonishyna YV, Ortner NJ, Kaserer T, Hoffmann J, Biskup S, Dafotakis M, et al. (February 2022). "Novel CACNA1A Variant p.Cys256Phe Disrupts Disulfide Bonds and Causes Spinocerebellar Ataxia". Movement Disorders. 37 (2). Movement disorders: official journal of the Movement Disorder Society: 401–404. doi:10.1002/mds.28835. PMID 34647648. S2CID 238859984.
External links
[edit]- Online Mendelian Inheritance in Man (OMIM): Spinocerebellar Ataxia, Autosomal Recessive 1; SCAR1 - 606002
- Online Mendelian Inheritance in Man (OMIM): Senataxin; SETX - 608465
- Ataxia and Cerebellar or Spinocerebellar Degeneration at NINDS
- Spinocerebellar Ataxias including Machado-Joseph Disease at NINDS
- Multiple System Atrophy at NINDS
- MedlinePlus Encyclopedia: Olivopontocerebellar atrophy
- Spinocerebellar ataxia 27 at NIH's Office of Rare Diseases
- Spinocerebellar ataxia dysmorphism at NIH's Office of Rare Diseases
| Authority control databases: National |
|---|