
Respiratory complex I: Difference between revisions
No edit summary |
No edit summary |
||
| Line 17: | Line 17: | ||
[[Image:NADH Dehydrogenase 2FUG Electron Carriers Labeled.png|thumb|235px|The electron carriers of the NADH dehydrogenease complex. Seven primary iron sulphur centers lie in a line down the peripheral arm of the complex to carry electrons from the site of NADH dehydration to [[ubiquinone]]. The iron sulphur group on the right is not found in the eukaryotic complex. Note: This image includes two errors. At the top, it should indicate NADH → NAD<sup>+</sup> via a FMN electron carrier/cofactor. At the bottom, it should indicate Ubiquinone → Ubiquinol.]] |
[[Image:NADH Dehydrogenase 2FUG Electron Carriers Labeled.png|thumb|235px|The electron carriers of the NADH dehydrogenease complex. Seven primary iron sulphur centers lie in a line down the peripheral arm of the complex to carry electrons from the site of NADH dehydration to [[ubiquinone]]. The iron sulphur group on the right is not found in the eukaryotic complex. Note: This image includes two errors. At the top, it should indicate NADH → NAD<sup>+</sup> via a FMN electron carrier/cofactor. At the bottom, it should indicate Ubiquinone → Ubiquinol.]] |
||
{{FixBunching|end}} |
{{FixBunching|end}} |
||
'''NADH dehydrogenase''' ({{EC number|1.6.5.3}}) is an [[enzyme]] located in the inner [[mitochondria|mitochondrial]] membrane that catalyzes the transfer of [[electron]]s from [[NADH]] to [[coenzyme Q]] (CoQ). It is |
'''NADH dehydrogenase''' ({{EC number|1.6.5.3}}) (also referred to as "NADH:quinone reductase" or "complex I") is an [[enzyme]] located in the inner [[mitochondria|mitochondrial]] membrane that catalyzes the transfer of [[electron]]s from [[NADH]] to [[coenzyme Q]] (CoQ). It is the "entry enzyme" of oxidative phosphorylation in the mitochondria. <ref name="pmid20074573">{{cite journal | author = Nakamaru-Ogiso E, Han H, Matsuno-Yagi A, Keinan E, Sinha SC, Yagi T, Ohnishi T. | title = The ND2 subunit is labeled by a photoaffinity analogue of asimicin, a potent complex I inhibitor. | journal = FEBS letters | volume = 584 | issue = | pages = 883-8 | year = 2010 | month = January | pmid = 20074573| doi = | url = | issn = }}</ref> |
||
==Function== |
==Function== |
||
| Line 31: | Line 31: | ||
All redox reactions take place in the extramembranous portion of NADH dehydrogenase. NADH initially binds to NADH dehydrogenase, and transfers two electrons to the flavin mononucleotide (FMN) prosthetic group of complex I, whose electron acceptor is identical to that of FAD – the isoalloxazine ring – creating FMNH2. The electrons are then transferred through the second prosthetic group of NADH dehydrogenase via a series of iron-sulfur (Fe-S) clusters, and finally to coenzyme Q (ubiquinone). This electron flow causes four hydrogen ions to be pumped out of the mitochondrial matrix. Quinone (Q) accepts two electrons to be reduce to ubiquional (QH2). <ref>{{cite book | author = Berg, J, Tymoczko, J, and L Stryer | title = Biochemistry | edition = 6th | language = | publisher = WH Freeman & Company | location = New York | year = 2006 | pages = 509-513 | }}</ref> |
All redox reactions take place in the extramembranous portion of NADH dehydrogenase. NADH initially binds to NADH dehydrogenase, and transfers two electrons to the flavin mononucleotide (FMN) prosthetic group of complex I, whose electron acceptor is identical to that of FAD – the isoalloxazine ring – creating FMNH2. The electrons are then transferred through the second prosthetic group of NADH dehydrogenase via a series of iron-sulfur (Fe-S) clusters, and finally to coenzyme Q (ubiquinone). This electron flow causes four hydrogen ions to be pumped out of the mitochondrial matrix. Quinone (Q) accepts two electrons to be reduce to ubiquional (QH2). <ref>{{cite book | author = Berg, J, Tymoczko, J, and L Stryer | title = Biochemistry | edition = 6th | language = | publisher = WH Freeman & Company | location = New York | year = 2006 | pages = 509-513 | }}</ref> |
||
{{FixBunching|beg}} |
|||
[[Image:NAD+toNADH.png|thumb|300px|left| NAD+ to NADH.]] |
[[Image:NAD+toNADH.png|thumb|300px|left| NAD+ to NADH.]] |
||
{{FixBunching|mid}} |
|||
[[Image:FMNtoFMNH2.png|thumb|300px|left| FMN to FMNH2.]] |
[[Image:FMNtoFMNH2.png|thumb|300px|left| FMN to FMNH2.]] |
||
{{FixBunching|mid}} |
|||
[[Image:QtoQH2.png|thumb|300px|left| Q to QH2.]] |
[[Image:QtoQH2.png|thumb|300px|left| Q to QH2.]] |
||
{{FixBunching|end}} |
|||
== Composition and structure == |
== Composition and structure == |
||
| Line 43: | Line 47: | ||
The structure is an "L" shape with a long membrane domain (with around 60 trans-membrane helices) and a hydrophilic peripheral domain, which includes all the known redox centres and the NADH binding site. Whereas the structure of the eukaryotic complex is not well characterised, the peripheral/hydrophilic domain of the complex from a bacterium (''Thermus thermophilus'') has been crystallised ({{PDB|2FUG}}).<ref name="pmid16469879">{{cite journal | author = Sazanov LA, Hinchliffe P | title = Structure of the hydrophilic domain of respiratory complex I from Thermus thermophilus | journal = Science | volume = 311 | issue = 5766 | pages = 1430–6 | year = 2006 | month = March | pmid = 16469879 | doi = 10.1126/science.1123809 | url = | issn = }}</ref> |
The structure is an "L" shape with a long membrane domain (with around 60 trans-membrane helices) and a hydrophilic peripheral domain, which includes all the known redox centres and the NADH binding site. Whereas the structure of the eukaryotic complex is not well characterised, the peripheral/hydrophilic domain of the complex from a bacterium (''Thermus thermophilus'') has been crystallised ({{PDB|2FUG}}).<ref name="pmid16469879">{{cite journal | author = Sazanov LA, Hinchliffe P | title = Structure of the hydrophilic domain of respiratory complex I from Thermus thermophilus | journal = Science | volume = 311 | issue = 5766 | pages = 1430–6 | year = 2006 | month = March | pmid = 16469879 | doi = 10.1126/science.1123809 | url = | issn = }}</ref> |
||
A recent by Roessler et al. (2010) used electron paramagnetic resonance (EPR) spectra and double electron-electron resonance (DEER) to determine the structure of complex I and the path of electron transfer through the iron-sulfur complexes. The iron-sulfur clusters are located in the hydrophilic domain. Seven of these clusters form a chain from the flavin to the quinone binding sites; the eighth cluster in on the other side of the flavin, and its function is unknown. The EPR and DEER results suggest an alternating or “roller-coaster” potential energy profile for the electron transfer between the active sites and along the iron-sulfur clusters, which can optimize the rate of electron travel and allow efficient energy conversion in complex I. <ref name="pmid20133838">{{cite journal | author = Roessler MM, King MS, Robinson AJ, Armstrong FA, Harmer J, Hirst J. | title = Direct assignment of EPR spectra to structurally defined iron-sulfur clusters in complex I by double electron-electron resonance.. | journal = Proceedings of the National Academy of Sciences of the United States of America | volume = 107 | issue = | pages = 1930-5 | year = 2010 | month = February | pmid = 20133838 | doi = | url = | issn = }}</ref> |
|||
==Inhibitors== |
==Inhibitors== |
||
| Line 48: | Line 55: | ||
Despite more than 50 years of study of NADH:ubiquinone oxidoreductase, no inhibitors blocking the electron flow inside the enzyme have been found. Hydrophobic inhibitors like rotenone or piericidin most likely disrupt the electron transfer between the terminal FeS cluster N2 and ubiquinone. Enzyme is also blocked by [[adenosine diphosphate ribose]] - reversible [[competitive inhibitor]] of NADH oxidation by the enzyme at the nucleotide binding site. Both hydrophylic NADH and hydrophobic ubiquinone analogs act at the beginning and the end of the internal electron-transport pathway respectively. |
Despite more than 50 years of study of NADH:ubiquinone oxidoreductase, no inhibitors blocking the electron flow inside the enzyme have been found. Hydrophobic inhibitors like rotenone or piericidin most likely disrupt the electron transfer between the terminal FeS cluster N2 and ubiquinone. Enzyme is also blocked by [[adenosine diphosphate ribose]] - reversible [[competitive inhibitor]] of NADH oxidation by the enzyme at the nucleotide binding site. Both hydrophylic NADH and hydrophobic ubiquinone analogs act at the beginning and the end of the internal electron-transport pathway respectively. |
||
The [[acetogenin]] family are the most potent complex I inhibitors. They have been shown to crosslink to the ND2 subunit, which suggests that ND2 is essential for quinone-binding. <ref name="pmid20074573">{{cite journal | author = Nakamaru-Ogiso E, Han H, Matsuno-Yagi A, Keinan E, Sinha SC, Yagi T, Ohnishi T. | title = The ND2 subunit is labeled by a photoaffinity analogue of asimicin, a potent complex I inhibitor. | journal = FEBS letters | volume = 584 | issue = | pages = 883-8 | year = 2010 | month = January | pmid = 20074573| doi = | url = | issn = }}</ref> Interestingly, Rolliniastatin-2, an acetogenin, is the first complex I inhibitor found that does not share the same binding site as rotenone. <ref name="pmid 8037664">{{cite journal | author = Degli Esposti M, Ghelli A, Ratta M, Cortes D, Estornell E. | title = Natural substances (acetogenins) from the family Annonaceae are powerful inhibitors of mitochondrial NADH dehydrogenase (Complex I). | journal = The Biochemical Journal | volume = 301 | issue = | pages = 161-7 | year = 1994 | month = July | pmid = 8037664| doi = | url = | issn = }}</ref> |
|||
==Active/de-active transition== |
==Active/de-active transition== |
||
| Line 62: | Line 72: | ||
During reverse electron transfer, Complex I might be the most important site of superoxide production within mitochondria, with up to 5% of electrons being diverted to superoxide formation. Reverse electron transfer, the process by which electrons from the reduced ubiquinol pool (supplied by [[succinate dehydrogenase]], [[glycerol-3-phosphate dehydrogenase]], or [[dihydro-oorotate dehydrogenase]] in mammalian mitochondria) pass through Complex I to reduce NAD<sup>+</sup> to NADH, driven by the inner mitochondrial membrane potential electric potential. Although it is not precisely known under what pathological conditions [[reverse-electron transfer]] would occur in vivo, in vitro experiments indicate that it can be a very potent source of superoxide when [[succinate]] concentrations are high and [[oxaloacetate]] or [[malate]] concentrations are low.<ref name="pmid17916065">{{cite journal | author = Muller FL, Liu Y, Abdul-Ghani MA, Lustgarten MS, Bhattacharya A, Jang YC, Van Remmen H | title = High rates of superoxide production in skeletal-muscle mitochondria respiring on both complex I- and complex II-linked substrates | journal = Biochem. J. | volume = 409 | issue = 2 | pages = 491–9 | year = 2008 | month = January | pmid = 17916065 | doi = 10.1042/BJ20071162 | url = | issn = }}</ref> |
During reverse electron transfer, Complex I might be the most important site of superoxide production within mitochondria, with up to 5% of electrons being diverted to superoxide formation. Reverse electron transfer, the process by which electrons from the reduced ubiquinol pool (supplied by [[succinate dehydrogenase]], [[glycerol-3-phosphate dehydrogenase]], or [[dihydro-oorotate dehydrogenase]] in mammalian mitochondria) pass through Complex I to reduce NAD<sup>+</sup> to NADH, driven by the inner mitochondrial membrane potential electric potential. Although it is not precisely known under what pathological conditions [[reverse-electron transfer]] would occur in vivo, in vitro experiments indicate that it can be a very potent source of superoxide when [[succinate]] concentrations are high and [[oxaloacetate]] or [[malate]] concentrations are low.<ref name="pmid17916065">{{cite journal | author = Muller FL, Liu Y, Abdul-Ghani MA, Lustgarten MS, Bhattacharya A, Jang YC, Van Remmen H | title = High rates of superoxide production in skeletal-muscle mitochondria respiring on both complex I- and complex II-linked substrates | journal = Biochem. J. | volume = 409 | issue = 2 | pages = 491–9 | year = 2008 | month = January | pmid = 17916065 | doi = 10.1042/BJ20071162 | url = | issn = }}</ref> |
||
Superoxide is a reactive oxygen species that contributes to cellular oxidative stress and is linked to neuromuscular diseases and aging. <ref name="pmid16682634">{{cite journal | author = Esterházy D, King MS, Yakovlev G, Hirst J. | title = Production of reactive oxygen species by complex I (NADH:ubiquinone oxidoreductase) from Escherichia coli and comparison to the enzyme from mitochondria. | journal = Biochemistry| volume = 25 | issue = | pages = 3964-71 | year = 2008 | month = March | pmid = 16682634 | doi = | url = | issn = }}</ref> NADH dehdyrogenase produces superoxide by transferring one electron from FMNH2 to oxygen (O2). The radical flavin leftover is unstable, and transfers the remaining electron to the iron-sulfur centres. Interestingly, it is the ratio of NADH to AND+ that determines the rate of superoxide formation. <ref name="pmid16682634">{{cite journal | author = Kussmaul L, Hirst J. | title = The mechanism of superoxide production by NADH:ubiquinone oxidoreductase (complex I) from bovine heart mitochondria. | journal = Proceedings of the National Academy of Sciences of the United States of America | volume = 103 | issue = | pages = 7607-12 | year = 2006 | month = May | pmid = 16682634 | doi = | url = | issn = }}</ref> |
|||
==Pathology== |
==Pathology== |
||
Mutations in the subunits of complex I can cause [[mitochondrial diseases]], including [[Leigh syndrome]]. Point mutations in various complex I subunits derived from mitochondrial DNA ([[mtDNA]]) can also result in [[Leber's Hereditary Optic Neuropathy]]. There is some evidence that complex I defects may play a role in the etiology of [[Parkinson's disease]], perhaps because of reactive oxygen species (complex I can, like [[complex III]], leak electrons to oxygen, forming highly toxic [[superoxide]]). |
Mutations in the subunits of complex I can cause [[mitochondrial diseases]], including [[Leigh syndrome]]. Point mutations in various complex I subunits derived from mitochondrial DNA ([[mtDNA]]) can also result in [[Leber's Hereditary Optic Neuropathy]]. There is some evidence that complex I defects may play a role in the etiology of [[Parkinson's disease]], perhaps because of reactive oxygen species (complex I can, like [[complex III]], leak electrons to oxygen, forming highly toxic [[superoxide]]). |
||
Although the exact etiology of Parkinson’s disease is unclear, it is likely that mitochondrial dysfunction, along with proteasome inhibition and environmental toxins, may play a large role. In fact, the inhibition of complex I has been shown to cause the production of peroxides and a decrease in proteasome activity, which may lead to Parkinson’s disease. <ref name="pmid20417232">{{cite journal | author = Chou AP, Li S, Fitzmaurice AG, Bronstein JM. | title = Mechanisms of rotenone-induced proteasome inhibition | journal = Neurotoxicology | volume = 113 | issue = | pages = 674-82 | year = 2010 | month = April | pmid = 20417232 | doi = | url = | issn = }}</ref> Additionally, Esteves et al. (2010) found that cell lines with Parkinson’s disease show increased proton leakage, which causes decreased maximum respiratory capacity. <ref name="pmid20132468">{{cite journal | author = Esteves AR, Lu J, Rodova M, Onyango I, Lezi E, Dubinsky R, Lyons KE, Pahwa R, Burns JM, Cardoso SM, Swerdlow RH. | title = Mitochondrial respiration and respiration-associated proteins in cell lines created through Parkinson's subject mitochondrial transfer. | journal = Journal of Neurochemistry | volume = 113 | issue = | pages = 674-82 | year = 2010 | month = February | pmid = 20132468| doi = | url = | issn = }}</ref> |
|||
Recent studies have examined other roles of NADH dehydrogenase activity in the brain. Andreazza et al. (2010) found that the level of complex I activity was significantly decreased in patients with bipolar disorder, but not in patients with depression or schizophrenia. They found that patients with bipolar disorder showed increased protein oxidation and nitration in their prefrontal cortex. These results suggest that future studies should target complex I for potential therapeutic studies for bipolar disorder. <ref name="pmid20368511">{{cite journal | author = Andreazza AC, Shao L, Wang JF, Young LT. | title = Mitochondrial complex I activity and oxidative damage to mitochondrial proteins in the prefrontal cortex of patients with bipolar disorder.| journal = Archives of General Psychiatry | volume = 67 | issue = | pages = 360-8 | year = 2010 | month = April | pmid = 20368511| doi = | url = | issn = }}</ref> Similarly, Moran et al. (2010) found that patients with severe complex I deficiency showed decreased oxygen consumption rates and slow growth rates. However, they found that mutations in different genes in complex I lead to different phenotypes, thereby explaining the variations of pathophysiological manifestatiosn of complex I deficiency. <ref name="pmid20153825">{{cite journal | author = Moran M, Rivera H, Sánchez-Aragó M, Blázquez A, Merinero B, Ugalde C, Arenas J, Cuezva JM, Martín MA. | title = Mitochondrial bioenergetics and dynamics interplay in complex I-deficient fibroblasts. | journal = Biochimica et Biophysica Acta | volume = 1802 | issue = | pages = 443-53 | year = 2010 | month = May | pmid = 20153825 | doi = | url = | issn = }}</ref> |
|||
Exposure to pesticides can also inhibit complex I and cause disease symptoms. For example, chronic exposure to low levels dichlorvos, an organophosphate used as a pesticide, has been shown to cause liver dysfunction. This occurs because dichlorvos alters complex I and II activity levels, which leads to decreased mitochondrial electron transfer activities and decreased ATP synthesis. <ref name="pmid 20132858">{{cite journal | author = Binukumar BK, Bal A, Kandimalla R, Sunkaria A, Gill KD. | title = Mitochondrial energy metabolism impairment and liver dysfunction following chronic exposure to dichlorvos. | journal = Toxicology | volume = 270 | issue = | pages = 77-84 | year = 2010 | month = April | pmid = 20132858| doi = | url = | issn = }}</ref> |
|||
==Genes== |
==Genes== |
||
Revision as of 05:26, 17 May 2010
| NADH dehydrogenase (ubiquinone) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Identifiers | |||||||||
| EC no. | 1.6.5.3 | ||||||||
| CAS no. | 9028-04-0 | ||||||||
| Databases | |||||||||
| IntEnz | IntEnz view | ||||||||
| BRENDA | BRENDA entry | ||||||||
| ExPASy | NiceZyme view | ||||||||
| KEGG | KEGG entry | ||||||||
| MetaCyc | metabolic pathway | ||||||||
| PRIAM | profile | ||||||||
| PDB structures | RCSB PDB PDBe PDBsum | ||||||||
| Gene Ontology | AmiGO / QuickGO | ||||||||
| |||||||||
{kind=link}


Template:FixBunching NADH dehydrogenase (EC 1.6.5.3) (also referred to as "NADH:quinone reductase" or "complex I") is an enzyme located in the inner mitochondrial membrane that catalyzes the transfer of electrons from NADH to coenzyme Q (CoQ). It is the "entry enzyme" of oxidative phosphorylation in the mitochondria. [1]
Function
NADH Dehydrogenase is the first enzyme (complex I) of the mitochondrial electron transport chain. There are three energy-transducing enzymes in the electron transport chain - NADH dehydrogenase (complex I), coenzyme Q (complex III), and cytochrome c oxidase (complex IV). [2] NADH dehydrogenase is the largest and most complicated enzyme of the electron transport chain.[3].
The reaction of NADH dehydrogenase is:
- NADH + H+ + CoQ + 4H+in → NAD+ + CoQH2 + 4H+out
In this process, the complex translocates 4 protons across the inner membrane per molecule of oxidized NADH, helping to build the electrochemical potential used to produce ATP. The reaction is reversible (NAD+ can be reduced by e.g. succinate in the presence of a high membrane potential) and the exact catalytic mechanism remains unknown.
Mechanism
All redox reactions take place in the extramembranous portion of NADH dehydrogenase. NADH initially binds to NADH dehydrogenase, and transfers two electrons to the flavin mononucleotide (FMN) prosthetic group of complex I, whose electron acceptor is identical to that of FAD – the isoalloxazine ring – creating FMNH2. The electrons are then transferred through the second prosthetic group of NADH dehydrogenase via a series of iron-sulfur (Fe-S) clusters, and finally to coenzyme Q (ubiquinone). This electron flow causes four hydrogen ions to be pumped out of the mitochondrial matrix. Quinone (Q) accepts two electrons to be reduce to ubiquional (QH2). [4]

{kind=link}

Composition and structure

NADH Dehydrogenase is the largest of the respiratory complexes, the mammalian enzyme containing 45 separate polypeptide chains. Of particular functional importance are the flavin prosthetic group and eight iron-sulfur clusters (FeS). Of the 45 subunits, seven are encoded by the mitochondrial genome.[5][6]
The structure is an "L" shape with a long membrane domain (with around 60 trans-membrane helices) and a hydrophilic peripheral domain, which includes all the known redox centres and the NADH binding site. Whereas the structure of the eukaryotic complex is not well characterised, the peripheral/hydrophilic domain of the complex from a bacterium (Thermus thermophilus) has been crystallised (PDB: 2FUG).[7]
A recent by Roessler et al. (2010) used electron paramagnetic resonance (EPR) spectra and double electron-electron resonance (DEER) to determine the structure of complex I and the path of electron transfer through the iron-sulfur complexes. The iron-sulfur clusters are located in the hydrophilic domain. Seven of these clusters form a chain from the flavin to the quinone binding sites; the eighth cluster in on the other side of the flavin, and its function is unknown. The EPR and DEER results suggest an alternating or “roller-coaster” potential energy profile for the electron transfer between the active sites and along the iron-sulfur clusters, which can optimize the rate of electron travel and allow efficient energy conversion in complex I. [8]
Inhibitors
The best-known inhibitor of complex I is rotenone (used as an organic pesticide). Rotenone and rotenoids are isoflavonoids occurring in several genera of tropical plants such as Antonia (Loganiaceae), Derris and Lonchocarpus (Faboideae, Fabaceae). There have been reports of rotenone-containing plants used by Indians to fish due to its ichthyotoxic effect, as early as the 17th century.[9] Rotenone binds to the ubiquinone binding site of Complex I as well as piericidin A another potent inhibitor with a close structural homologue to ubiquinone.
Despite more than 50 years of study of NADH:ubiquinone oxidoreductase, no inhibitors blocking the electron flow inside the enzyme have been found. Hydrophobic inhibitors like rotenone or piericidin most likely disrupt the electron transfer between the terminal FeS cluster N2 and ubiquinone. Enzyme is also blocked by adenosine diphosphate ribose - reversible competitive inhibitor of NADH oxidation by the enzyme at the nucleotide binding site. Both hydrophylic NADH and hydrophobic ubiquinone analogs act at the beginning and the end of the internal electron-transport pathway respectively.
The acetogenin family are the most potent complex I inhibitors. They have been shown to crosslink to the ND2 subunit, which suggests that ND2 is essential for quinone-binding. [1] Interestingly, Rolliniastatin-2, an acetogenin, is the first complex I inhibitor found that does not share the same binding site as rotenone. [10]
Active/de-active transition
The catalytic properties of eukaryotic complex I are not simple. Two catalytically and structurally distinct forms exist in any given preparation of the enzyme: one is the fully competent, so-called “active” A-form and the other is the catalytically silent, dormant, “de-activated”, D-form. After exposure of idle enzyme to elevated, but physiological temperatures (>30°C) in the absence of substrate, the enzyme converts to the D-form. This form is catalytically incompetent but can be activated by the slow reaction (k~4 min-1) of NADH oxidation with subsequent ubiquinone reduction. After one or several turnovers the enzyme becomes active and can catalyse physiological NADH:ubiquinone reaction at a much higher rate (k~104 min-1). In the presence of divalent cations (Mg2+, Ca2+), or at alkaline pH the activation takes much longer.
The high activation energy (270 kJ/mol) of the deactivation process indicates the occurrence of major conformational changes in the organisation of the Complex I. However, until now, the only conformational difference observed between these two forms is the number of cysteine residues exposed at the surface of the enzyme. Treatment of the D-form of complex I with the sulfhydryl reagents N-Ethylmaleimide or DTNB irreversibly blocks critical cysteine residue(s), abolishing the ability of the enzyme to respond to activation, thus inactivating it irreversibly. The A-form of complex I is insensitive to sulfhydryl reagents.
It was found that these conformational changes may have a very important physiological significance. The de-active, but not the active form of Complex I was susceptible to inhibition by nitrosothiols and peroxynitrite.[11] It is likely that transition from the active to the deactive form of complex I takes place during pathological conditions when the turnover of the enzyme is limited at physiological temperatures, such as during hypoxia, or when the tissue nitric oxide:oxygen ratio increases (i.e. metabolic hypoxia).[12]
Production of superoxide
Recent investigations suggest that Complex I is a potent source of reactive oxygen species.[13] Complex I can produce superoxide (as well as hydrogen peroxide), through at least two different pathways. During forward electron transfer, only very small amounts of superoxide are produced (probably less than 0.1% of the overall electron flow).[13][14]
During reverse electron transfer, Complex I might be the most important site of superoxide production within mitochondria, with up to 5% of electrons being diverted to superoxide formation. Reverse electron transfer, the process by which electrons from the reduced ubiquinol pool (supplied by succinate dehydrogenase, glycerol-3-phosphate dehydrogenase, or dihydro-oorotate dehydrogenase in mammalian mitochondria) pass through Complex I to reduce NAD+ to NADH, driven by the inner mitochondrial membrane potential electric potential. Although it is not precisely known under what pathological conditions reverse-electron transfer would occur in vivo, in vitro experiments indicate that it can be a very potent source of superoxide when succinate concentrations are high and oxaloacetate or malate concentrations are low.[15]
Superoxide is a reactive oxygen species that contributes to cellular oxidative stress and is linked to neuromuscular diseases and aging. [16] NADH dehdyrogenase produces superoxide by transferring one electron from FMNH2 to oxygen (O2). The radical flavin leftover is unstable, and transfers the remaining electron to the iron-sulfur centres. Interestingly, it is the ratio of NADH to AND+ that determines the rate of superoxide formation. [16]
Pathology
Mutations in the subunits of complex I can cause mitochondrial diseases, including Leigh syndrome. Point mutations in various complex I subunits derived from mitochondrial DNA (mtDNA) can also result in Leber's Hereditary Optic Neuropathy. There is some evidence that complex I defects may play a role in the etiology of Parkinson's disease, perhaps because of reactive oxygen species (complex I can, like complex III, leak electrons to oxygen, forming highly toxic superoxide).
Although the exact etiology of Parkinson’s disease is unclear, it is likely that mitochondrial dysfunction, along with proteasome inhibition and environmental toxins, may play a large role. In fact, the inhibition of complex I has been shown to cause the production of peroxides and a decrease in proteasome activity, which may lead to Parkinson’s disease. [17] Additionally, Esteves et al. (2010) found that cell lines with Parkinson’s disease show increased proton leakage, which causes decreased maximum respiratory capacity. [18]
Recent studies have examined other roles of NADH dehydrogenase activity in the brain. Andreazza et al. (2010) found that the level of complex I activity was significantly decreased in patients with bipolar disorder, but not in patients with depression or schizophrenia. They found that patients with bipolar disorder showed increased protein oxidation and nitration in their prefrontal cortex. These results suggest that future studies should target complex I for potential therapeutic studies for bipolar disorder. [19] Similarly, Moran et al. (2010) found that patients with severe complex I deficiency showed decreased oxygen consumption rates and slow growth rates. However, they found that mutations in different genes in complex I lead to different phenotypes, thereby explaining the variations of pathophysiological manifestatiosn of complex I deficiency. [20]
Exposure to pesticides can also inhibit complex I and cause disease symptoms. For example, chronic exposure to low levels dichlorvos, an organophosphate used as a pesticide, has been shown to cause liver dysfunction. This occurs because dichlorvos alters complex I and II activity levels, which leads to decreased mitochondrial electron transfer activities and decreased ATP synthesis. [21]
Genes
The following is a list of humans genes that encode components of the NADH dehydrogenase (ubiquinone) complex:
- NADH dehydrogenase (ubiquinone) 1 alpha subcomplex
- NDUFA1 – NADH dehydrogenase (ubiquinone) 1 alpha subcomplex, 1, 7.5kDa
- NDUFA2 – NADH dehydrogenase (ubiquinone) 1 alpha subcomplex, 2, 8kDa
- NDUFA3 – NADH dehydrogenase (ubiquinone) 1 alpha subcomplex, 3, 9kDa
- NDUFA4 – NADH dehydrogenase (ubiquinone) 1 alpha subcomplex, 4, 9kDa
- NDUFA4L – NADH dehydrogenase (ubiquinone) 1 alpha subcomplex, 4-like
- NDUFA4L2 – NADH dehydrogenase (ubiquinone) 1 alpha subcomplex, 4-like 2
- NDUFA5 – NADH dehydrogenase (ubiquinone) 1 alpha subcomplex, 5, 13kDa
- NDUFA6 – NADH dehydrogenase (ubiquinone) 1 alpha subcomplex, 6, 14kDa
- NDUFA7 – NADH dehydrogenase (ubiquinone) 1 alpha subcomplex, 7, 14.5kDa
- NDUFA8 – NADH dehydrogenase (ubiquinone) 1 alpha subcomplex, 8, 19kDa
- NDUFA9 – NADH dehydrogenase (ubiquinone) 1 alpha subcomplex, 9, 39kDa
- NDUFA10 – NADH dehydrogenase (ubiquinone) 1 alpha subcomplex, 10, 42kDa
- NDUFA11 – NADH dehydrogenase (ubiquinone) 1 alpha subcomplex, 11, 14.7kDa
- NDUFA12 – NADH dehydrogenase (ubiquinone) 1 alpha subcomplex, 12
- NDUFA13 – NADH dehydrogenase (ubiquinone) 1 alpha subcomplex, 13
- NDUFAB1 – NADH dehydrogenase (ubiquinone) 1, alpha/beta subcomplex, 1, 8kDa
- NDUFAF1 – NADH dehydrogenase (ubiquinone) 1 alpha subcomplex, assembly factor 1
- NDUFAF2 – NADH dehydrogenase (ubiquinone) 1 alpha subcomplex, assembly factor 2
- NDUFAF3 – NADH dehydrogenase (ubiquinone) 1 alpha subcomplex, assembly factor 3
- NDUFAF4 – NADH dehydrogenase (ubiquinone) 1 alpha subcomplex, assembly factor 4
- NADH dehydrogenase (ubiquinone) 1 beta subcomplex
- NDUFB1 – NADH dehydrogenase (ubiquinone) 1 beta subcomplex, 1, 7kDa
- NDUFB2 – NADH dehydrogenase (ubiquinone) 1 beta subcomplex, 2, 8kDa
- NDUFB3 – NADH dehydrogenase (ubiquinone) 1 beta subcomplex, 3, 12kDa
- NDUFB4 – NADH dehydrogenase (ubiquinone) 1 beta subcomplex, 4, 15kDa
- NDUFB5 – NADH dehydrogenase (ubiquinone) 1 beta subcomplex, 5, 16kDa
- NDUFB6 – NADH dehydrogenase (ubiquinone) 1 beta subcomplex, 6, 17kDa
- NDUFB7 – NADH dehydrogenase (ubiquinone) 1 beta subcomplex, 7, 18kDa
- NDUFB8 – NADH dehydrogenase (ubiquinone) 1 beta subcomplex, 8, 19kDa
- NDUFB9 – NADH dehydrogenase (ubiquinone) 1 beta subcomplex, 9, 22kDa
- NDUFB10 – NADH dehydrogenase (ubiquinone) 1 beta subcomplex, 10, 22kDa
- NDUFB11 – NADH dehydrogenase (ubiquinone) 1 beta subcomplex, 11, 17.3kDa
- NADH dehydrogenase (ubiquinone) 1, subcomplex unknown
- NADH dehydrogenase (ubiquinone) Fe-S protein
- NDUFS1 – NADH dehydrogenase (ubiquinone) Fe-S protein 1, 75kDa (NADH-coenzyme Q reductase)
- NDUFS2 – NADH dehydrogenase (ubiquinone) Fe-S protein 2, 49kDa (NADH-coenzyme Q reductase)
- NDUFS3 – NADH dehydrogenase (ubiquinone) Fe-S protein 3, 30kDa (NADH-coenzyme Q reductase)
- NDUFS4 – NADH dehydrogenase (ubiquinone) Fe-S protein 4, 18kDa (NADH-coenzyme Q reductase)
- NDUFS5 – NADH dehydrogenase (ubiquinone) Fe-S protein 5, 15kDa (NADH-coenzyme Q reductase)
- NDUFS6 – NADH dehydrogenase (ubiquinone) Fe-S protein 6, 13kDa (NADH-coenzyme Q reductase)
- NDUFS7 – NADH dehydrogenase (ubiquinone) Fe-S protein 7, 20kDa (NADH-coenzyme Q reductase)
- NDUFS8 – NADH dehydrogenase (ubiquinone) Fe-S protein 8, 23kDa (NADH-coenzyme Q reductase)
- NADH dehydrogenase (ubiquinone) flavoprotein 1
- mitochondrially encoded NADH dehydrogenase subunit
- MT-ND1 - mitochondrially encoded NADH dehydrogenase subunit 1
- MT-ND2 - mitochondrially encoded NADH dehydrogenase subunit 2
- MT-ND3 - mitochondrially encoded NADH dehydrogenase subunit 3
- MT-ND4 - mitochondrially encoded NADH dehydrogenase subunit 4
- MT-ND4L - mitochondrially encoded NADH dehydrogenase subunit 4L
- MT-ND5 - mitochondrially encoded NADH dehydrogenase subunit 5
- MT-ND6 - mitochondrially encoded NADH dehydrogenase subunit 6
References
- ^ a b Nakamaru-Ogiso E, Han H, Matsuno-Yagi A, Keinan E, Sinha SC, Yagi T, Ohnishi T. (2010). "The ND2 subunit is labeled by a photoaffinity analogue of asimicin, a potent complex I inhibitor". FEBS letters. 584: 883–8. PMID 20074573.
{{cite journal}}: Unknown parameter|month=ignored (help)CS1 maint: multiple names: authors list (link) - ^ Berg, J, Tymoczko, J, and L Stryer (2006). Biochemistry (6th ed.). New York: WH Freeman & Company. pp. 509–513.
{{cite book}}: Cite has empty unknown parameter:|1=(help)CS1 maint: multiple names: authors list (link) - ^ Brandt, U (2006). "Energy converting NADH:quinone oxidoreductase (complex I)". Annual Review of Biochemistry. 75: 69–92. doi:10.1146/annurev.biochem.75.103004.142539.
{{cite journal}}: Cite has empty unknown parameter:|month=(help) - ^ Berg, J, Tymoczko, J, and L Stryer (2006). Biochemistry (6th ed.). New York: WH Freeman & Company. pp. 509–513.
{{cite book}}: Cite has empty unknown parameter:|1=(help)CS1 maint: multiple names: authors list (link) - ^ Voet, Judith G.; Voet, Donald (2004). Biochemistry (3rd ed.). New York: J. Wiley & Sons. p. 813–826. ISBN 0-471-19350-X.
{{cite book}}: CS1 maint: multiple names: authors list (link) - ^ Carroll J, Fearnley IM, Skehel JM, Shannon RJ, Hirst J, Walker JE (2006). "Bovine complex I is a complex of 45 different subunits". J. Biol. Chem. 281 (43): 32724–7. doi:10.1074/jbc.M607135200. PMID 16950771.
{{cite journal}}: Unknown parameter|month=ignored (help)CS1 maint: multiple names: authors list (link) CS1 maint: unflagged free DOI (link) - ^ Sazanov LA, Hinchliffe P (2006). "Structure of the hydrophilic domain of respiratory complex I from Thermus thermophilus". Science. 311 (5766): 1430–6. doi:10.1126/science.1123809. PMID 16469879.
{{cite journal}}: Unknown parameter|month=ignored (help) - ^ Roessler MM, King MS, Robinson AJ, Armstrong FA, Harmer J, Hirst J. (2010). "Direct assignment of EPR spectra to structurally defined iron-sulfur clusters in complex I by double electron-electron resonance.". Proceedings of the National Academy of Sciences of the United States of America. 107: 1930–5. PMID 20133838.
{{cite journal}}: Unknown parameter|month=ignored (help)CS1 maint: multiple names: authors list (link) - ^ Moretti C, Grenand P (1982). "[The "nivrées", or ichthyotoxic plants of French Guyana]". J Ethnopharmacol (in French). 6 (2): 139–60. PMID 7132401.
{{cite journal}}: Unknown parameter|month=ignored (help) - ^ Degli Esposti M, Ghelli A, Ratta M, Cortes D, Estornell E. (1994). "Natural substances (acetogenins) from the family Annonaceae are powerful inhibitors of mitochondrial NADH dehydrogenase (Complex I)". The Biochemical Journal. 301: 161–7. PMID 8037664.
{{cite journal}}: Unknown parameter|month=ignored (help)CS1 maint: multiple names: authors list (link) - ^ Galkin A, Moncada S (2007). "S-nitrosation of mitochondrial complex I depends on its structural conformation". J. Biol. Chem. 282 (52): 37448–53. doi:10.1074/jbc.M707543200. PMID 17956863.
{{cite journal}}: Unknown parameter|month=ignored (help)CS1 maint: unflagged free DOI (link) - ^ Moncada S, Erusalimsky JD (2002). "Does nitric oxide modulate mitochondrial energy generation and apoptosis?". Nat. Rev. Mol. Cell Biol. 3 (3): 214–20. doi:10.1038/nrm762. PMID 11994742.
{{cite journal}}: Unknown parameter|month=ignored (help) - ^ a b Murphy MP (2009). "How mitochondria produce reactive oxygen species". Biochem. J. 417 (1): 1–13. doi:10.1042/BJ20081386. PMC 2605959. PMID 19061483.
{{cite journal}}: Unknown parameter|month=ignored (help) - ^ Hansford RG, Hogue BA, Mildaziene V (1997). "Dependence of H2O2 formation by rat heart mitochondria on substrate availability and donor age". J. Bioenerg. Biomembr. 29 (1): 89–95. PMID 9067806.
{{cite journal}}: Unknown parameter|month=ignored (help)CS1 maint: multiple names: authors list (link) - ^ Muller FL, Liu Y, Abdul-Ghani MA, Lustgarten MS, Bhattacharya A, Jang YC, Van Remmen H (2008). "High rates of superoxide production in skeletal-muscle mitochondria respiring on both complex I- and complex II-linked substrates". Biochem. J. 409 (2): 491–9. doi:10.1042/BJ20071162. PMID 17916065.
{{cite journal}}: Unknown parameter|month=ignored (help)CS1 maint: multiple names: authors list (link) - ^ a b Esterházy D, King MS, Yakovlev G, Hirst J. (2008). "Production of reactive oxygen species by complex I (NADH:ubiquinone oxidoreductase) from Escherichia coli and comparison to the enzyme from mitochondria". Biochemistry. 25: 3964–71. PMID 16682634.
{{cite journal}}: Unknown parameter|month=ignored (help)CS1 maint: multiple names: authors list (link) Cite error: The named reference "pmid16682634" was defined multiple times with different content (see the help page). - ^ Chou AP, Li S, Fitzmaurice AG, Bronstein JM. (2010). "Mechanisms of rotenone-induced proteasome inhibition". Neurotoxicology. 113: 674–82. PMID 20417232.
{{cite journal}}: Unknown parameter|month=ignored (help)CS1 maint: multiple names: authors list (link) - ^ Esteves AR, Lu J, Rodova M, Onyango I, Lezi E, Dubinsky R, Lyons KE, Pahwa R, Burns JM, Cardoso SM, Swerdlow RH. (2010). "Mitochondrial respiration and respiration-associated proteins in cell lines created through Parkinson's subject mitochondrial transfer". Journal of Neurochemistry. 113: 674–82. PMID 20132468.
{{cite journal}}: Unknown parameter|month=ignored (help)CS1 maint: multiple names: authors list (link) - ^ Andreazza AC, Shao L, Wang JF, Young LT. (2010). "Mitochondrial complex I activity and oxidative damage to mitochondrial proteins in the prefrontal cortex of patients with bipolar disorder". Archives of General Psychiatry. 67: 360–8. PMID 20368511.
{{cite journal}}: Unknown parameter|month=ignored (help)CS1 maint: multiple names: authors list (link) - ^ Moran M, Rivera H, Sánchez-Aragó M, Blázquez A, Merinero B, Ugalde C, Arenas J, Cuezva JM, Martín MA. (2010). "Mitochondrial bioenergetics and dynamics interplay in complex I-deficient fibroblasts". Biochimica et Biophysica Acta. 1802: 443–53. PMID 20153825.
{{cite journal}}: Unknown parameter|month=ignored (help)CS1 maint: multiple names: authors list (link) - ^ Binukumar BK, Bal A, Kandimalla R, Sunkaria A, Gill KD. (2010). "Mitochondrial energy metabolism impairment and liver dysfunction following chronic exposure to dichlorvos". Toxicology. 270: 77–84. PMID 20132858.
{{cite journal}}: Unknown parameter|month=ignored (help)CS1 maint: multiple names: authors list (link)
Additional images
-
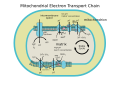 ETC
ETC
External links
- Interactive Molecular model of NADH dehydrogenase (Requires MDL Chime)
- Complex I home page at The Scripps Research Institute
- Electron+Transport+Complex+I at the U.S. National Library of Medicine Medical Subject Headings (MeSH)
- NADH+Dehydrogenase at the U.S. National Library of Medicine Medical Subject Headings (MeSH)
 | This EC 1.6 enzyme-related article is a stub. You can help Wikipedia by expanding it. |