
Congenital hypothyroidism
This article needs additional citations for verification. (May 2013) |
| Congenital hypothyroidism | |
|---|---|
| Specialty | Endocrinology |

Congenital hypothyroidism (CH) is a condition of thyroid hormone deficiency present at birth. Approximately 1 in 4000 newborn infants has a severe deficiency of thyroid function, while even more have mild or partial degrees. If untreated for several months after birth, severe congenital hypothyroidism can lead to growth failure and permanent intellectual disability. Treatment consists of a daily dose of thyroid hormone (thyroxine) by mouth. Because the treatment is simple, effective, and inexpensive, nearly all of the developed world practices newborn screening to detect and treat congenital hypothyroidism in the first weeks of life.
Signs and symptoms
Infants born with congenital hypothyroidism may show no effects, or may display mild effects that often go unrecognized as a problem: excessive sleeping, reduced interest in nursing, poor muscle tone, low or hoarse cry, infrequent bowel movements, exaggerated jaundice, and low body temperature. If fetal deficiency was severe because of complete absence (athyreosis) of the gland, physical features may include a larger anterior fontanel, persistence of a posterior fontanel, an umbilical hernia, and a large tongue (macroglossia).
In the era before newborn screening, less than half of cases of severe hypothyroidism were recognized in the first month of life. As the months proceeded, these infants would grow poorly and be delayed in their development. By several years of age, they would display the recognizable facial and body features of cretinism. Persistence of severe, untreated hypothyroidism resulted in severe mental impairment, with an IQ below 80 in the majority. Most of these children eventually ended up in institutional care.
-
 3 month old infant with untreated CH; picture demonstrates hypotonic posture, myxedematous facies, macroglossia, and umbilical hernia
3 month old infant with untreated CH; picture demonstrates hypotonic posture, myxedematous facies, macroglossia, and umbilical hernia -
 Close up of face, showing myxedematous facies, macroglossia, and skin mottling
Close up of face, showing myxedematous facies, macroglossia, and skin mottling -
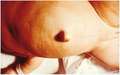 Close up showing abdominal distension and umbilical hernia.
Close up showing abdominal distension and umbilical hernia.



Cause
Around the world, the most common cause of congenital hypothyroidism is iodine deficiency, but in most of the developed world and areas of adequate environmental iodine, cases are due to a combination of known and unknown causes. Most commonly there is a defect of development of the thyroid gland itself, resulting in an absent (athyreosis) or underdeveloped (hypoplastic) gland. A hypoplastic gland may develop higher in the neck or even in the back of the tongue. A gland in the wrong place is referred to as ectopic, and an ectopic gland at the base or back of the tongue is a lingual thyroid. Some of these cases of developmentally abnormal glands result from genetic defects, and some are "sporadic," with no identifiable cause. One Japanese study found a statistical correlation between certain organochlorine insecticides and dioxin-like chemicals in the milk of mothers who had given birth to infants with congenital hypothyroidism.[1]
In some instances, hypothyroidism detected by screening may be transient. One common cause of this is the presence of maternal antibodies that temporarily impair thyroid function for several weeks.[2]
Cretinism is an old term for the state of mental and physical retardation resulting from untreated congenital hypothyroidism, usually due to iodine deficiency from birth because of low iodine levels in the soil and local food sources. The term, like so many other 19th century medical terms, acquired pejorative connotations as it became used in lay speech. It is now rarely used by physicians.
Genetic
Congenital hypothyroidism can also occur due to genetic defects of thyroxine or triiodothyronine synthesis within a structurally normal gland. Among specific defects are thyrotropin (TSH) resistance, iodine trapping defect, organification defect, thyroglobulin, and iodotyrosine deiodinase deficiency. In a small proportion of cases of congenital hypothyroidism, the defect is due to a deficiency of thyroid stimulating hormone, either isolated or as part of congenital hypopituitarism.
Genetic types of nongoitrous congenital hypothyroidism include:
| OMIM | Name | Gene |
|---|---|---|
| Template:OMIM2 | congenital hypothyroidism, nongoitrous 1 CHNG1 | TSHR |
| Template:OMIM2 | CHNG2 | PAX8 |
| Template:OMIM2 | CHNG3 | ? at 15q25.3-q26.1 |
| Template:OMIM2 | CHNG4 | TSHB |
| Template:OMIM2 | CHNG5 | NKX2-5 |
Nongoitrous congenital hypothyroidism has been described as the "most prevalent inborn endocrine disorder".[3]
Diagnosis
In the developed world, nearly all cases of congenital hypothyroidism are detected by the newborn screening program. These are based on measurement of TSH or thyroxine (T4) on the second or third day of life (Heel prick).
If the TSH is high, or the T4 low, the infant's doctor and parents are called and a referral to a pediatric endocrinologist is recommended to confirm the diagnosis and initiate treatment. Often a technetium (Tc-99m pertechnetate) thyroid scan is performed to detect a structurally abnormal gland. A radioactive iodine (RAIU) exam will help differentiate congenital absence or a defect in organification (a process necessary to make thyroid hormone).
Treatment
The goal of newborn screening programs is to detect and start treatment within the first 1–2 weeks of life. Treatment consists of a daily dose of thyroxine, available as a small tablet. The generic name is levothyroxine, and several brands are available. Commonly used brands in North America are Synthroid, Levoxyl, Unithroid, and Levothroid. The tablet is crushed and given to the infant with a small amount of water or milk. The most commonly recommended dose range is 10-15 μg/kg daily, typically 12.5 to 37.5 or 44 μg.[4] Within a few weeks, the T4 and TSH levels are rechecked to confirm that they are being normalized by treatment. As the child grows up, these levels are checked regularly to maintain the right dose. The dose increases as the child grows.
Prognosis
Most children born with congenital hypothyroidism and correctly treated with thyroxine grow and develop normally in all respects. Even most of those with athyreosis and undetectable T4 levels at birth develop with normal intelligence, although as a population academic performance tends to be below that of siblings and mild learning problems occur in some.[5]
Congenital hypothyroidism is the most common preventable cause of intellectual disability. Few treatments in the practice of medicine provide as large a benefit for as small an effort.
The developmental quotient (DQ, as per Gesell Developmental Schedules) of children with hypothyroidism at age 24 months that have received treatment within the first 3 weeks of birth is summarised below:
| . | Adaptive behavior | Fine motor | Gross motor | Language | Personal-social behavior |
|---|---|---|---|---|---|
| Severe CH | 92 | 89 | 90 | 89 | 90 |
| Moderate CH | 97 | 97 | 98 | 96 | 96 |
| Mild CH | 100 | 99 | 100 | 99 | 100 |
Epidemiology
Neonatal thyroid screening programs from all over the world have revealed that congenital hypothyroidism (CH) occurs with an incidence of 1:3000 to 1:4000.[7][8][9][10] The differences in CH-incidence are more likely due to iodine deficiency thyroid disorders or to the type of screening method than to ethnic affiliation.[7] CH is caused by an absent or defective thyroid gland classified into agenesis (22-42%), ectopy (35-42%) and gland in place defects (24-36%).[7][11] It is also found to be of increased association with female sex and gestational age >40 weeks.[11]
References
- ^ Nagayama J, Kohno H, Kunisue T, et al. (2007). "Concentrations of organochlorine pollutants in mothers who gave birth to neonates with congenital hypothyroidism". Chemosphere. 68 (5): 972–6. doi:10.1016/j.chemosphere.2007.01.010. PMID 17307219.
- ^ "Congenital hypothyroidism". Orphanet. August 2010. Retrieved 22 May 2012.
- ^ Grasberger H, Vaxillaire M, Pannain S, et al. (December 2005). "Identification of a locus for nongoitrous congenital hypothyroidism on chromosome 15q25.3-26.1". Hum. Genet. 118 (3–4): 348–55. doi:10.1007/s00439-005-0036-6. PMID 16189712.
- ^ LaFranchi SH, Austin J (2007). "How should we be treating children with congenital hypothyroidism?". J. Pediatr. Endocrinol. Metab. 20 (5): 559–78. doi:10.1515/JPEM.2007.20.5.559. PMID 17642417.
- ^ Moltz KC, Postellon DC (1994). "Congenital hypothyroidism and mental development". Compr Ther. 20 (6): 342–6. PMID 8062543.
- ^ Huo K, Zhang Z, Zhao D, Li H, Wang J, Wang X, Feng H, Wang X, Zhu C (2011). "Risk factors for neurodevelopmental deficits in congenital hypothyroidism after early substitution treatment". Endocrine. 58 (5): 355–61. doi:10.1507/endocrj.k10e-384.
- ^ a b c Klett, M (1997). "Epidemiology of congenital hypothyroidism". Experimental and Clinical Endocrinology & Diabetes. 105 Suppl 4: 19–23. doi:10.1055/s-0029-1211926. PMID 9439909.
- ^ Harris, KB; Pass, KA (July 2007). "Increase in congenital hypothyroidism in New York State and in the United States". Molecular genetics and metabolism. 91 (3): 268–77. doi:10.1016/j.ymgme.2007.03.012. PMID 17512233.
- ^ Deladoey, J.; Belanger, N.; Van Vliet, G. (1 August 2007). "Random Variability in Congenital Hypothyroidism from Thyroid Dysgenesis over 16 Years in Quebec". Journal of Clinical Endocrinology & Metabolism. 92 (8): 3158–3161. doi:10.1210/jc.2007-0527. PMID 17504897.
- ^ Olney, RS; Grosse, SD; Vogt RF, Jr (May 2010). "Prevalence of congenital hypothyroidism--current trends and future directions: workshop summary". Pediatrics. 125 Suppl 2: S31-6. doi:10.1542/peds.2009-1975C. PMID 20435715.
- ^ a b Medda, E; Olivieri, A; Stazi, MA; Grandolfo, ME; Fazzini, C; Baserga, M; Burroni, M; Cacciari, E; Calaciura, F; Cassio, A; Chiovato, L; Costa, P; Leonardi, D; Martucci, M; Moschini, L; Pagliardini, S; Parlato, G; Pignero, A; Pinchera, A; Sala, D; Sava, L; Stoppioni, V; Tancredi, F; Valentini, F; Vigneri, R; Sorcini, M (December 2005). "Risk factors for congenital hypothyroidism: results of a population case-control study (1997-2003)". European Journal of Endocrinology. 153 (6): 765–73. doi:10.1530/eje.1.02048. PMID 16322381.
This template is no longer used; please see Template:Endocrine pathology for a suitable replacement