
Glycogen branching enzyme
| GBE1 | |||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||
| Identifiers | |||||||||||||||||||||||||||||||||||||||||||||||||||
| Aliases | GBE1, APBD, GBE, GSD4, glucan (1,4-alpha-), branching enzyme 1, 1,4-alpha-glucan branching enzyme 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||
| External IDs | OMIM: 607839 MGI: 1921435 HomoloGene: 129 GeneCards: GBE1 | ||||||||||||||||||||||||||||||||||||||||||||||||||
| EC number | 2.4.1.18 | ||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||
| Wikidata | |||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||




| Glycogen branching enzyme | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Identifiers | |||||||||
| EC no. | 2.4.1.18 | ||||||||
| CAS no. | 9001-97-2 | ||||||||
| Databases | |||||||||
| IntEnz | IntEnz view | ||||||||
| BRENDA | BRENDA entry | ||||||||
| ExPASy | NiceZyme view | ||||||||
| KEGG | KEGG entry | ||||||||
| MetaCyc | metabolic pathway | ||||||||
| PRIAM | profile | ||||||||
| PDB structures | RCSB PDB PDBe PDBsum | ||||||||
| Gene Ontology | AmiGO / QuickGO | ||||||||
| |||||||||
1,4-alpha-glucan-branching enzyme, also known as brancher enzyme or glycogen-branching enzyme is an enzyme that in humans is encoded by the GBE1 gene.[5]
Glycogen branching enzyme is an enzyme that adds branches to the growing glycogen molecule during the synthesis of glycogen, a storage form of glucose. More specifically, during glycogen synthesis, a glucose 1-phosphate molecule reacts with uridine triphosphate (UTP) to become UDP-glucose, an activated form of glucose. The activated glucosyl unit of UDP-glucose is then transferred to the hydroxyl group at the C-4 of a terminal residue of glycogen to form an α-1,4-glycosidic linkage, a reaction catalyzed by glycogen synthase. Importantly, glycogen synthase can only catalyze the synthesis of α-1,4-glycosidic linkages. Since glycogen is a readily mobilized storage form of glucose, the extended glycogen polymer is branched by glycogen branching enzyme to provide glycogen breakdown enzymes, such as glycogen phosphorylase, with many terminal residues for rapid degradation. Branching also importantly increases the solubility and decreases the osmotic strength of glycogen.[6]
The protein encoded by this gene is a glycogen branching enzyme that catalyzes the transfer of alpha-1,4-linked glucosyl units from the outer end of a glycogen chain to an alpha-1,6 position on the same or a neighboring glycogen chain. Branching of the chains is essential to increase the solubility of the glycogen molecule and, consequently, in reducing the osmotic pressure within cells. The highest levels of this enzyme are found in liver and muscle cells. Mutations in this gene are associated with glycogen storage disease type IV (also known as Andersen's disease).
Nomenclature[edit]
This enzyme belongs to the family of transferases, to be specific, those glycosyltransferases that transfer hexoses (hexosyltransferases). The systematic name of this enzyme class is 1,4-alpha-D-glucan:1,4-alpha-D-glucan 6-alpha-D-(1,4-alpha-D-glucano)-transferase. Other names in common use include branching enzyme, amylo-(1,4→1,6)-transglycosylase, Q-enzyme, alpha-glucan-branching glycosyltransferase, amylose isomerase, enzymatic branching factor, branching glycosyltransferase, enzyme Q, glucosan transglycosylase, 1,4-alpha-glucan branching enzyme 1, plant branching enzyme, alpha-1,4-glucan:alpha-1,4-glucan-6-glycosyltransferase, and starch branching enzyme. This enzyme participates in starch and sucrose metabolism.
Gene[edit]
GBE is encoded by the GBE1 gene.[5][7][8][9]
Through Southern blot analysis of DNA derived from human/rodent somatic cell hybrids, GBE1 has been identified as an autosomal gene located on the short arm of chromosome 3 at position 12.3.[7][8][9][10] The human GBE gene was also isolated by a function complementation of the Saccharomyces cerevisiae GBE deficiency.[10] From the isolated cDNA, the length of the gene was found to be approximately 3 kb.[10] Additionally, the coding sequence was found to comprise 2,106 base pairs and encode a 702-amino acid long GBE. The molecular mass of human GBE was calculated to be 80,438 Da.[10]
Structure[edit]

Glycogen branching enzyme belongs to the α-amylase family of enzymes, which include α-amylases, pullulanas/isoamylase, cyclodextrin glucanotransferase (CGT), and branching enzyme.[11][12] Shown by x-ray crystallography, glycogen branching enzyme has four marginally asymmetric units each that are organized into three domains: an amino-terminal domain, involved in determining the length of the chain transfer, a carboxyl-terminal domain, involved in substrate preference and catalytic capacity, and a central (α/β) barrel catalytic domain.[11][13][14][15] The amino-terminal domain consists of 128 residues arranged in seven β-strands, the carboxyl-terminal domain with 116 residues also organized in seven β-strands, and the (α/β) barrel domain with 372 residues. While the central (α/β) barrel domain is common in members of the α-amylase family, numerous variations exist between the various barrel domains. Additionally, there are striking differences between the loops connecting elements of the secondary structure among these various α-amylase members, especially around the active site. In comparison to the other family members, glycogen binding enzyme has shorter loops, which result in a more open cavity, favorable to the binding of a bulkier substrate such as branched sugar. Through primary structure analysis and the x-ray crystallographic structures of the members of the α-amylase family, seven residue were conserved, Asp335, His340, Arg403, Asp 405, Glu458, His525, and Asp526 (E coli. numbering). These residues are implicated in catalysis and substrate binding.[11]
Glycogen binding enzymes in other organisms have also been crystallized and structurally determined, demonstrating both similarity and variation to GBE found in Escherichia coli.[16][17][18][19]
Function[edit]
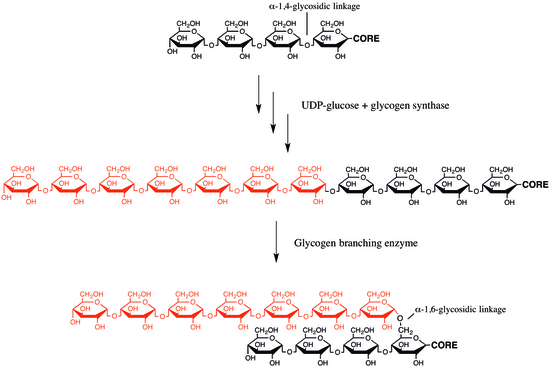
In glycogen, every 10 to 14 glucose units, a side branch with an additional chain of glucose units occurs. The side chain attaches at carbon atom 6 of a glucose unit, an α-1,6-glycosidic bond. This connection is catalyzed by a branching enzyme, generally given the name α-glucan branching enzyme. A branching enzyme attaches a string of seven glucose units (with some minor variation to this number) to the carbon at the C-6 position on the glucose unit, forming the α-1,6-glycosidic bond. The specific nature of this enzyme means that this chain of 7 carbons is usually attached to a glucose molecule that is in position three from the non-reducing end of another chain. Because the enzyme works with such specificity regarding the number of glucose units transferred and the position to which they are transferred, the enzyme creates the very characteristic, highly branched glycogen molecule.[20]
Clinical significance[edit]
Mutations in this gene are associated with glycogen storage disease type IV (also known as Andersen's disease) in newborns and with adult polyglucosan body disease.[5][21]
Approximately 40 mutations in the GBE1 gene, most resulting in a point mutation in the glycogen branching enzyme, have led to the early childhood disorder, glycogen storage disease type IV (GSD IV).[9] This disease is characterized by a severe depletion or complete absence of GBE, resulting in the accumulation of abnormally structured glycogen, known as polyglucosan bodies.[9] Glycogen buildup leads to increased osmotic pressure resulting in cellular swelling and death.[9] The tissues most affected by this disease are the liver, heart, and neuromuscular system, areas with the greatest levels of glycogen accumulation.[9][22] Abnormal glycogen buildup in the liver interferes with liver functioning and can result in an enlarged liver and liver disease.[9][23] In muscles, the inability of cells to efficiently breakdown glycogen due to the severe reduction or absence of branching can lead to muscle weakness and atrophy.[9] At least three mutations in the GBE1 gene have been found to cause another disease called adult polyglucosan body disease (APBD).[9][24] While in GSD IV GBE activity is undetectable or minimally detectable, APBD is characterized by reduced or even normal GBE activity.[24] In this disease, abnormal glycogen can build up in neurons leading to a spectrum of problems. Specifically, some disease characteristics are gait difficulties from mixed upper and lower motor neuron involvement sensory loss in lower extremities, and neurogenic bladder, a problem in which a person lacks bladder control due to a brain, spinal cord, or nerve condition.[24][25]
References[edit]
- ^ a b c GRCh38: Ensembl release 89: ENSG00000114480 – Ensembl, May 2017
- ^ a b c GRCm38: Ensembl release 89: ENSMUSG00000022707 – Ensembl, May 2017
- ^ "Human PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- ^ "Mouse PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- ^ a b c "Entrez Gene: glucan (1,4-alpha-), branching enzyme 1". Retrieved 2011-08-30.
- ^ Berg J (2012). Biochemistry Seventh Edition. W.H. Freeman and Company. pp. 627–630.
- ^ a b National Center for Biotechnology Information. "GBE1 glucan (1,4-alpha-), branching enzyme 1 [ Homo sapiens (human) ]". US. National Library of Medicine.
- ^ a b Online Mendelian Inheritance in Man. "Glycogen Branching Enzyme; GBE1". Johns Hopkins University.
- ^ a b c d e f g h i Genetics Home Reference. "GBE1". U.S. National Library of Medicine.
- ^ a b c d Thon VJ, Khalil M, Cannon JF (April 1993). "Isolation of human glycogen branching enzyme cDNAs by screening complementation in yeast". The Journal of Biological Chemistry. 268 (10): 7509–13. doi:10.1016/S0021-9258(18)53204-X. PMID 8463281.
- ^ a b c Abad MC, Binderup K, Rios-Steiner J, Arni RK, Preiss J, Geiger JH (November 2002). "The X-ray crystallographic structure of Escherichia coli branching enzyme". The Journal of Biological Chemistry. 44. 277 (44): 42164–70. doi:10.1074/jbc.m205746200. PMID 12196524.
- ^ Pal K, Kumar S, Sharma S, Garg SK, Alam MS, Xu HE, Agrawal P, Swaminathan K (July 2010). "Crystal structure of full-length Mycobacterium tuberculosis H37Rv glycogen branching enzyme: insights of N-terminal beta-sandwich in substrate specificity and enzymatic activity". The Journal of Biological Chemistry. 285 (27): 20897–903. doi:10.1074/jbc.M110.121707. PMC 2898361. PMID 20444687.
- ^ Matsuura Y, Kusunoki M, Harada W, Kakudo M (March 1984). "Structure and possible catalytic residues of Taka-amylase A". Journal of Biochemistry. 95 (3): 697–702. doi:10.1093/oxfordjournals.jbchem.a134659. PMID 6609921.
- ^ Buisson G, Duée E, Haser R, Payan F (December 1987). "Three dimensional structure of porcine pancreatic alpha-amylase at 2.9 A resolution. Role of calcium in structure and activity". The EMBO Journal. 6 (13): 3909–16. doi:10.1002/j.1460-2075.1987.tb02731.x. PMC 553868. PMID 3502087.
- ^ Devillers CH, Piper ME, Ballicora MA, Preiss J (October 2003). "Characterization of the branching patterns of glycogen branching enzyme truncated on the N-terminus". Archives of Biochemistry and Biophysics. 418 (1): 34–8. doi:10.1016/S0003-9861(03)00341-2. PMID 13679080.
- ^ Kuriki T, Stewart DC, Preiss J (November 1997). "Construction of chimeric enzymes out of maize endosperm branching enzymes I and II: activity and properties". The Journal of Biological Chemistry. 272 (46): 28999–9004. doi:10.1074/jbc.272.46.28999. PMID 9360973.
- ^ Palomo M, Pijning T, Booiman T, Dobruchowska JM, van der Vlist J, Kralj S, Planas A, Loos K, Kamerling JP, Dijkstra BW, van der Maarel MJ, Dijkhuizen L, Leemhuis H (February 2011). "Thermus thermophilus glycoside hydrolase family 57 branching enzyme: crystal structure, mechanism of action, and products formed". The Journal of Biological Chemistry. 286 (5): 3520–30. doi:10.1074/jbc.m110.179515. PMC 3030357. PMID 21097495.
- ^ Santos CR, Tonoli CC, Trindade DM, Betzel C, Takata H, Kuriki T, Kanai T, Imanaka T, Arni RK, Murakami MT (February 2011). "Structural basis for branching-enzyme activity of glycoside hydrolase family 57: structure and stability studies of a novel branching enzyme from the hyperthermophilic archaeon Thermococcus kodakaraensis KOD1" (PDF). Proteins. 79 (2): 547–57. doi:10.1002/prot.22902. PMID 21104698. S2CID 25862890.
- ^ Noguchi J, Chaen K, Vu NT, Akasaka T, Shimada H, Nakashima T, Nishi A, Satoh H, Omori T, Kakuta Y, Kimura M (August 2011). "Crystal structure of the branching enzyme I (BEI) from Oryza sativa L with implications for catalysis and substrate binding". Glycobiology. 21 (8): 1108–16. doi:10.1093/glycob/cwr049. PMID 21493662.
- ^ Rose S (1999). The Chemistry of Life. Pelican Books. pp. 199–201.
- ^ McKusick VA, Kniffin CL (May 2, 2016). "OMIM Entry 263570 - Polyglucosan body neuropathy, adult form". Online Mendelian Inheritance in Man. Johns Hopkins University. Retrieved 7 March 2017.
- ^ Mingyi, Chen (2011). Glycogen Storages Diseases chapter of Molecular Pathology of Liver Diseases. Springer. pp. 677–682. ISBN 9781441971074.
- ^ Bruno C, van Diggelen OP, Cassandrini D, Gimpelev M, Giuffrè B, Donati MA, Introvini P, Alegria A, Assereto S, Morandi L, Mora M, Tonoli E, Mascelli S, Traverso M, Pasquini E, Bado M, Vilarinho L, van Noort G, Mosca F, DiMauro S, Zara F, Minetti C (September 2004). "Clinical and genetic heterogeneity of branching enzyme deficiency (glycogenosis type IV)". Neurology. 63 (6): 1053–8. doi:10.1212/01.wnl.0000138429.11433.0d. PMID 15452297. S2CID 7874969.
- ^ a b c Klein, Christopher (1993). "GBE1 Adult Polyglucosan Body Disease". Adult Polyglucosan Body Disease. University of Washington, Seattle. GBE1 Adult Polyglucosan Body Disease. PMID 20301758.
{{cite book}}:|journal=ignored (help) - ^ Hussain A, Armistead J, Gushulak L, Kruck C, Pind S, Triggs-Raine B, Natowicz MR (September 2012). "The adult polyglucosan body disease mutation GBE1 c.1076A>C occurs at high frequency in persons of Ashkenazi Jewish background". Biochemical and Biophysical Research Communications. 426 (2): 286–8. doi:10.1016/j.bbrc.2012.08.089. PMID 22943850.
Further reading[edit]
- Barker SA, Bourne E, Peat S (1949). "The enzymic synthesis and degradation of starch. Part IV. The purification and storage of the Q-enzyme of the potato". Journal of the Chemical Society (Resumed): 1705–1711. doi:10.1039/jr9490001705.
- Baum H, Gilbert GA (May 1953). "A simple method for the preparation of crystalline potato phosphorylase and Q-enzyme". Nature. 171 (4361): 983–4. Bibcode:1953Natur.171..983B. doi:10.1038/171983a0. PMID 13063502. S2CID 2243647.
- Hehre EJ (1951). "Enzymic Synthesis of Polysaccharides: A Biological type of Polymerization". Advances in Enzymology and Related Areas of Molecular Biology. Advances in Enzymology - and Related Areas of Molecular Biology. Vol. 11. pp. 297–337. doi:10.1002/9780470122563.ch6. ISBN 978-0-470-12256-3. PMID 24540594.
{{cite book}}:|journal=ignored (help) - Allan CM, Wang Y, Jimenez M, Marshan B, Spaliviero J, Illingworth P, Handelsman DJ (March 2006). "Follicle-stimulating hormone increases primordial follicle reserve in mature female hypogonadal mice". The Journal of Endocrinology. 188 (3): 549–57. doi:10.1677/joe.1.06614. PMID 16522734.
- Stelzl U, Worm U, Lalowski M, Haenig C, Brembeck FH, Goehler H, Stroedicke M, Zenkner M, Schoenherr A, Koeppen S, Timm J, Mintzlaff S, Abraham C, Bock N, Kietzmann S, Goedde A, Toksöz E, Droege A, Krobitsch S, Korn B, Birchmeier W, Lehrach H, Wanker EE (September 2005). "A human protein-protein interaction network: a resource for annotating the proteome". Cell. 122 (6): 957–68. doi:10.1016/j.cell.2005.08.029. hdl:11858/00-001M-0000-0010-8592-0. PMID 16169070. S2CID 8235923.
- Massa R, Bruno C, Martorana A, de Stefano N, van Diggelen OP, Federico A (April 2008). "Adult polyglucosan body disease: proton magnetic resonance spectroscopy of the brain and novel mutation in the GBE1 gene". Muscle & Nerve. 37 (4): 530–6. doi:10.1002/mus.20916. PMID 17994551. S2CID 18749059.
- Rose JE, Behm FM, Drgon T, Johnson C, Uhl GR (2010). "Personalized smoking cessation: interactions between nicotine dose, dependence and quit-success genotype score". Molecular Medicine. 16 (7–8): 247–53. doi:10.2119/molmed.2009.00159. PMC 2896464. PMID 20379614.
- Hannula-Jouppi K, Kaminen-Ahola N, Taipale M, Eklund R, Nopola-Hemmi J, Kääriäinen H, Kere J (October 2005). "The axon guidance receptor gene ROBO1 is a candidate gene for developmental dyslexia". PLOS Genetics. 1 (4): e50. doi:10.1371/journal.pgen.0010050. PMC 1270007. PMID 16254601.
- Konstantinidou AE, Anninos H, Dertinger S, Nonni A, Petersen M, Karadimas C, Havaki S, Marinos E, Akman HO, DiMauro S, Patsouris E (April 2008). "Placental involvement in glycogen storage disease type IV". Placenta. 29 (4): 378–81. doi:10.1016/j.placenta.2008.01.005. PMID 18289670.
- Melén E, Himes BE, Brehm JM, Boutaoui N, Klanderman BJ, Sylvia JS, Lasky-Su J (September 2010). "Analyses of shared genetic factors between asthma and obesity in children". The Journal of Allergy and Clinical Immunology. 126 (3): 631–7.e1–8. doi:10.1016/j.jaci.2010.06.030. PMC 2941152. PMID 20816195.
- Bruno C, van Diggelen OP, Cassandrini D, Gimpelev M, Giuffrè B, Donati MA, Introvini P, Alegria A, Assereto S, Morandi L, Mora M, Tonoli E, Mascelli S, Traverso M, Pasquini E, Bado M, Vilarinho L, van Noort G, Mosca F, DiMauro S, Zara F, Minetti C (September 2004). "Clinical and genetic heterogeneity of branching enzyme deficiency (glycogenosis type IV)". Neurology. 63 (6): 1053–8. doi:10.1212/01.wnl.0000138429.11433.0d. PMID 15452297. S2CID 7874969.
- Ziemssen F, Sindern E, Schröder JM, Shin YS, Zange J, Kilimann MW, Malin JP, Vorgerd M (April 2000). "Novel missense mutations in the glycogen-branching enzyme gene in adult polyglucosan body disease". Annals of Neurology. 47 (4): 536–40. doi:10.1002/1531-8249(200004)47:4<536::AID-ANA22>3.0.CO;2-K. PMID 10762170. S2CID 42856760.
- McCarthy JJ, Meyer J, Moliterno DJ, Newby LK, Rogers WJ, Topol EJ (December 2003). "Evidence for substantial effect modification by gender in a large-scale genetic association study of the metabolic syndrome among coronary heart disease patients". Human Genetics. 114 (1): 87–98. doi:10.1007/s00439-003-1026-1. PMID 14557872. S2CID 2568593.
- Tay SK, Akman HO, Chung WK, Pike MG, Muntoni F, Hays AP, Shanske S, Valberg SJ, Mickelson JR, Tanji K, DiMauro S (April 2004). "Fatal infantile neuromuscular presentation of glycogen storage disease type IV". Neuromuscular Disorders. 14 (4): 253–60. doi:10.1016/j.nmd.2003.12.006. PMID 15019703. S2CID 34056349.
- Pescador N, Villar D, Cifuentes D, Garcia-Rocha M, Ortiz-Barahona A, Vazquez S, Ordoñez A, Cuevas Y, Saez-Morales D, Garcia-Bermejo ML, Landazuri MO, Guinovart J, del Peso L (March 2010). "Hypoxia promotes glycogen accumulation through hypoxia inducible factor (HIF)-mediated induction of glycogen synthase 1". PLOS ONE. 5 (3): e9644. Bibcode:2010PLoSO...5.9644P. doi:10.1371/journal.pone.0009644. PMC 2837373. PMID 20300197.
- Bruno C, Cassandrini D, Assereto S, Akman HO, Minetti C, Di Mauro S (July 2007). "Neuromuscular forms of glycogen branching enzyme deficiency". Acta Myologica. 26 (1): 75–8. PMC 2949312. PMID 17915577.
- Flachsbart F, Franke A, Kleindorp R, Caliebe A, Blanché H, Schreiber S, Nebel A (December 2010). "Investigation of genetic susceptibility factors for human longevity - a targeted nonsynonymous SNP study". Mutation Research. 694 (1–2): 13–9. doi:10.1016/j.mrfmmm.2010.08.006. PMID 20800603.
- Rush J, Moritz A, Lee KA, Guo A, Goss VL, Spek EJ, Zhang H, Zha XM, Polakiewicz RD, Comb MJ (January 2005). "Immunoaffinity profiling of tyrosine phosphorylation in cancer cells". Nature Biotechnology. 23 (1): 94–101. doi:10.1038/nbt1046. PMID 15592455. S2CID 7200157.
- Bailey SD, Xie C, Do R, Montpetit A, Diaz R, Mohan V, Keavney B, Yusuf S, Gerstein HC, Engert JC, Anand S (October 2010). "Variation at the NFATC2 locus increases the risk of thiazolidinedione-induced edema in the Diabetes REduction Assessment with ramipril and rosiglitazone Medication (DREAM) study". Diabetes Care. 33 (10): 2250–3. doi:10.2337/dc10-0452. PMC 2945168. PMID 20628086.
External links[edit]
- GeneReviews/NCBI/NIH/UW entry on Adult Polyglucosan Body Disease
- OMIM entries on Adult Polyglucosan Body Disease
- Overview of all the structural information available in the PDB for UniProt: Q04446 (1,4-alpha-glucan-branching enzyme) at the PDBe-KB.