
Mitotic inhibitor

A mitotic inhibitor, microtubule inhibitor, or tubulin inhibitor, is a drug that inhibits mitosis, or cell division, and is used in treating cancer, gout, and nail fungus. These drugs disrupt microtubules, which are structures that pull the chromosomes apart when a cell divides. Mitotic inhibitors are used in cancer treatment, because cancer cells are able to grow through continuous division that eventually spread through the body (metastasize). Thus, cancer cells are more sensitive to inhibition of mitosis than normal cells. Mitotic inhibitors are also used in cytogenetics (the study of chromosomes), where they stop cell division at a stage where chromosomes can be easily examined.[1]
Mitotic inhibitors are derived from natural substances such as plant alkaloids, and prevent cells from undergoing mitosis by disrupting microtubule polymerization, thus preventing cancerous growth. Microtubules are long, ropelike proteins that extend through the cell and move cellular components around. Microtubules are long polymers made of smaller units (monomers) of the protein tubulin. Microtubules are created during normal cell functions by assembling (polymerizing) tubulin components, and are disassembled when they are no longer needed. One of the important functions of microtubules is to move and separate chromosomes and other components of the cell for cell division (mitosis). Mitotic inhibitors interfere with the assembly and disassembly of tubulin into microtubule polymers. This interrupts cell division, usually during the mitosis (M) phase of the cell cycle when two sets of fully formed chromosomes are supposed to separate into daughter cells.[2][3] Tubulin binding molecules have generated significant interest after the introduction of the taxanes into clinical oncology and the general use of the vinca alkaloids.
Examples of mitotic inhibitors frequently used in the treatment of cancer include paclitaxel, docetaxel, vinblastine, vincristine, and vinorelbine.[1] Colchicine and griseofulvin are mitotic inhibitors used in the treatment of gout and nail fungus, respectively.
Microtubules
[edit]
Microtubules are the key components of the cytoskeleton of eukaryotic cells and have an important role in various cellular functions such as intracellular migration and transport, cell shape maintenance, polarity, cell signaling and mitosis.[4] They play a critical role in cell division by involving in the movement and attachment of the chromosomes during various stages of mitosis. Therefore, microtubule dynamics is an important target for the developing anti-cancer drugs.[5]
Structure
[edit]Microtubules are composed of two globular protein subunits, α- and β-tubulin. These two subunits combine to form an α,β-heterodimer which then assembles in a filamentous tube-shaped structure. The tubulin hetero-dimers arrange themselves in a head to tail manner with the α-subunit of one dimer coming in contact with the β-subunit of the other. This arrangement results in the formation of long protein fibres called protofilaments.
These protofilaments form the backbone of the hollow, cylindrical microtubule which is about 25 nanometers in diameter and varies from 200 nanometers to 25 micrometers in length. About 12–13 protofilaments arrange themselves in parallel to form a C-shaped protein sheet, which then curls around to give a pipe-like structure called the microtubule. The head to tail arrangement of the hetero dimers gives polarity to the resulting microtubule, which has an α-subunit at one end and a β-subunit at the other end. The α-tubulin end has negative (–) charges while the β-tubulin end has positive (+) charges.[4] The microtubule grows from discrete assembly sites in the cells called Microtubule organizing centers (MTOCs), which are a network of microtubule associated proteins (MAP).[6][7]
Two molecules of energy rich guanosine triphosphate (GTP) are also important components of the microtubule structure. One molecule of GTP is tightly bound to the α-tubulin and is non-exchangeable whereas the other GTP molecule is bound to β-tubulin and can be easily exchanged with guanosine diphosphate (GDP). The stability of the microtubule will depend on whether the β-end is occupied by GTP or GDP. A microtubule having a GTP molecule at the β-end will be stable and continue to grow whereas a microtubule having a GDP molecule at the β-end will be unstable and will depolymerise rapidly.[6][7]
Microtubule dynamics
[edit]Microtubules are not static but they are highly dynamic polymers and exhibit two kinds of dynamic behaviors : 'dynamic instability' and 'treadmilling'. Dynamic instability is a process in which the microtubule ends switches between periods of growth and shortening. The two ends are not equal, the α-tubulin ringed (-)end is less dynamic while the more dynamic β-tubulin ringed (+) end grows and shortens more rapidly. Microtubule undergoes long periods of slow lengthening, brief periods of rapid shortening and also a pause in which there is neither growth nor shortening.[4][7][8] Dynamic instability is characterized by four variables: the rate of microtubule growth; the rate of shortening; frequency of transition from the growth or paused state to shortening (called a 'catastrophe') and the frequency of transition from shortening to growth or pause (called a 'rescue').
The other dynamic behavior called treadmilling is the net growth of the microtubule at one end and the net shortening at the other end. It involves the intrinsic flow of tubulin sub-units from the plus end to the minus end. Both the dynamic behaviors are important and a particular microtubule may exhibit primarily dynamic instability, treadmilling or a mixture of both.[8][9]
Mechanism of action
[edit]Agents which act as inhibitors of tubulin, also act as inhibitors of cell division. A microtubule exists in a continuous dynamic state of growing and shortening by reversible association and dissociation of α/β-tubulin heterodimers at both the ends. This dynamic behavior and resulting control over the length of the microtubule is vital to the proper functioning of the mitotic spindle in mitosis i.e., cell division.
Microtubule is involved in different stages of the cell cycle. During the first stage or prophase, the microtubules required for cell division begins to form and grow towards the newly formed chromosomes forming a bundle of microtubules called the mitotic spindle. During prometaphase and metaphase this spindle attaches itself to the chromosomes at a particular point called the kinetochore and undergoes several growing and shortening periods in tuning with the back and forth oscillations of the chromosomes. In anaphase also, the microtubules attached to the chromosomes maintain a carefully regulated shortening and lengthening process. Thus the presence of a drug which can suppress the microtubule dynamics is sufficient to block the cell cycle and result in the death of the cells by apoptosis.[5][10][11]
Tubulin inhibitors thus act by interfering with the dynamics of the microtubule, i.e., growing (polymerization) and shortening (depolymerization). One class of inhibitors operate by inhibiting polymerization of tubulin to form microtubules and are called polymerization inhibitors like the colchicine analogues and the vinca alkaloids. They decrease the microtubule polymer mass in the cells at high concentration and act as microtubule-destabilizing agents. The other class of inhibitors operate by inhibiting the depolymerization of polymerized tubulin and increases the microtubule polymer mass in the cells. They act as microtubule-stabilizing agents and are called depolymerization inhibitors like the paclitaxel analogues.[4] These three classes of drugs seems to operate by slightly different mechanism.

Colchicine analogues blocks cell division by disrupting the microtubule. It has been reported that the β-subunit of tubulin is involved in colchicine binding. It binds to the soluble tubulin to form colchicine-tubulin complex. This complex along with the normal tubulins then undergoes polymerization to form the microtubule. However the presence of this T-C complex prevents further polymerization of the microtubule . This complex brings about a conformational change which blocks the tubulin dimers from further addition and thereby prevents the growth of the microtubule. As the T-C complex slows down the addition of new dimers, the microtubule disassembles due to structural imbalance or instability during the metaphase of mitosis.[13]
The Vinca alkaloids bind to the β-subunit of tubulin dimers at a distinct region called the Vinca-binding domain. They bind to tubulin rapidly, and this binding is reversible and independent of temperature (between 0 °C and 37 °C). In contrast to colchicine, vinca alkaloids bind to the microtubule directly. They do not first form a complex with the soluble tubulin nor do they copolymerize to form the microtubule, however they are capable of bringing about a conformational change in tubulin in connection with tubulin self-association.[8] Vinca alkaloids bind to the tubulin with high affinity at the microtubule ends but with low affinity at the tubulin sites present along the sides of the microtubule cylinder. The binding of these drugs at the high affinity sites results in strong kinetic suppression of tubulin exchange even at low drug concentration while their binding to the low affinity sites in relatively high drug concentration depolymerizes microtubules.[5]
In contrast to colchicine and vinca alkaloids, paclitaxel enhances microtubule polymerization promoting both the nucleation and elongation phases of the polymerization reaction, and it reduces the critical tubulin sub-unit concentration (i.e., soluble tubulin concentration at steady- state). Microtubules polymerized in presence of paclitaxel are extremely stable.[5] The binding mechanism of the paclitaxel mimic that of the GTP nucleotide along with some important differences. GTP binds at one end of the tubulin dimer keeping contact with the next dimer along each of the protofilament while the paclitaxel binds to one side of β-tubulin keeping contact with the next protofilament. GTP binds to unassembled tubulin dimers whereas paclitaxel binding sites are located only in assembled tubulin. The hydrolysis of GTP permits the disassembly and the regulation of the microtubule system; however, the activation of tubulin by paclitaxel results in permanent stabilization of the microtubule. Thus the suppression of microtubule dynamics was described to be the main cause of the inhibition of cell division and of tumor cell death in paclitaxel treated cells.[14]
Structure activity relationship (SAR)
[edit]
Colchicine is one of the oldest known antimitotic drugs and in the past years[when?] much research has been done in order to isolate or develop compounds having similar structure but high activity and less toxicity. This resulted in the discovery of a number of colchicine analogues. The structure of colchicine is made up of three rings, a trimethoxy benzene ring (ring A), a methoxy tropone ring (ring C) and a seven-membered ring (ring B) with an acetamido group located at its C-7 position. The trimethoxy phenyl group of colchicine not only helps in stabilizing the tubulin-colchicine complex but is also important for antitubulin activity in conjunction with the ring C. The 3-methoxy group increased the binding ability whereas the 1-methoxy group helped in attaining the correct conformation of the molecule. The stability of the tropone ring and the position of the methoxy and carbonyl group are crucial for the binding ability of the compound. The 10-methoxy group can be replaced with halogen, alkyl, alkoxy or amino groups without affecting tubulin binding affinity, while bulky substituents reduce the activity. Ring B when expanded showed reduced activity, however the ring and its C-7 side chain is thought to affect the conformation of the colchicine analogues rather than their tubulin binding ability. Substitution at C-5 resulted in loss of activity whereas attachment of annulated heterocyclic ring systems to ring B resulted in highly potent compound.[13]

Paclitaxel has achieved great success as an anti-cancer drug, yet there has been continuous effort to improve its efficacy and develop analogues which are more active and have greater bioavailability and specificity. The importance of C-13 substituted phenylisoserine side chain to bioactivity of paclitaxel has been known for a long time. Several replacements at the C-3' substitution have been tested. Replacement of the C-3' phenyl group with alkyl or alkyneyl groups greatly enhanced the activity, and with CF3 group at that position in combination with modification of the 10-Ac with other acyl groups increased the activity several times. Another modification of C-3' with cyclopropane and epoxide moieties were also found to be potent. Most of the analogues without ring A were found to be much less active than paclitaxel itself. The analogues with amide side chain at C-13 are less active than their ester counterpart. Also deoxygenation at position 1 showed reduced activity. Preparation of 10-α-spiro epoxide and its 7-MOM ether gave compounds having comparable cytotoxicity and tubulin assembly activity as that of paclitaxel. Substitution with C-6-α-OH and C-6-β-OH gave analogues which were equipotent to paclitaxel in tubulin assembly assay. Finally the oxetane ring is found to play an important role during interaction with tubulin.[15]

Vinblastine is a highly potent drug which also has serious side effects especially on the neurological system. Therefore, new synthetic analogues were developed with the goal of obtaining more efficient and less toxic drugs. The stereochemical configurations at C-20', C-16' and C-14' in the velbanamine portion are critical and inversion leads to loss of activity. The C-16' carboxymethyl group is important for activity since decarboxylated dimer is inactive. Structural variation at C-15'- C-20' in the velbanamine ring is well tolerated. The upper skeletal modification of vinblastine gave vinorelbine which shows comparable activity as that of vinblastine. Another analogue prepared was the difluoro derivative of vinorelbine which showed improved in vivo antitumor activity. It was discovered that fluorination at C-19' position of vinorelbine dramatically increased the in vivo activity. Most of the SAR studies involve the vindoline portion of bis-indole alkaloids because modification at C-16 and C-17 offers good opportunities for developing new analogues. The replacement of the ester group with an amide group at the C-16 resulted in the development of vindesine. Similarly replacement of the acetyl group at C-16 with L-trp-OC2H5, d-Ala(P)-(OC2H5)2, L-Ala(P)-(OC2H5)2 and I-Vla(P)-(OC2H5)2 gave rise to new analogues having anti- tubulin activity. Also it was found that the vindoline's indole methyl group is a useful position to functionalize potentially and develop new, potent vinblastine derivatives. A new series of semi-synthetic C-16 -spiro-oxazolidine-1,3-diones prepared from 17-deacetyl vinblastine showed good anti-tubulin activity and lower cytotoxicity. Vinglycinate a glycinate prodrug derived from the C-17-OH group of vinblastine showed similar antitumor activity and toxicity as that of vinblastine.[16]
Use in cytogenetics
[edit]Cytogenetics, the study of chromosomal material by analysis of G-Banded chromosomes, uses mitotic inhibitors extensively. In order to prepare a slide for cytogenetic study, a mitotic inhibitor is added to the cells being studied. This stops the cells during mitosis, while the chromosomes are still visible. Once the cells are centrifuged and placed in a hypotonic solution, they swell, spreading the chromosomes. After preparation, the chromosomes of the cells can be viewed under a microscope to have the banding patterns of the chromosomes examined. This experiment is crucial to many forms of cancer research.
Tubulin binding drugs
[edit]Tubulin binding molecules differ from the other anticancer drugs in their mode of action because they target the mitotic spindle and not the DNA. Tubulin binding drugs have been classified on the basis of their mode of action and binding site[6][17][18] as:
I. Tubulin depolymerization inhibitors
[edit]a) Paclitaxel site ligands, includes the paclitaxel, epothilone, docetaxel, discodermolide etc.
II. Tubulin polymerization inhibitors
[edit]a) Colchicine binding site, includes the colchicine, combrestatin, 2-methoxyestradiol, methoxy benzenesulfonamides (E7010) etc.
b) Vinca alkaloids binding site,[19] includes vinblastine, vincristine, vinorelbine, vinflunine, dolastatins, halichondrins, hemiasterlins, cryptophysin 52, etc.
- Binding site of different drugs on tubulin
-
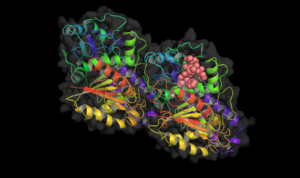 Taxol bound to tubulin.
Taxol bound to tubulin. -
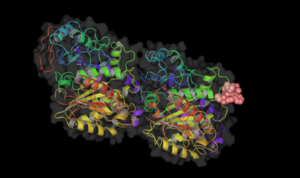 Vinblastine bound to tubulin.
Vinblastine bound to tubulin. -
 Colchicine bound to tubulin.
Colchicine bound to tubulin.



| Classes of tubulin inhibitors | Binding domain | Related drugs or analogs | Therapeutic uses | Stage of clinical development |
|---|---|---|---|---|
| Polymerization inhibitors | Vinca domain | Vinblastine | Hodgkin's disease, testicular germ cell cancer | in clinical use; 22 combination trials in progress |
| Vincristine | Leukemia, lymphomas | In clinical use; 108 combination trials in progress | ||
| Vinorelbine | Solid tumours, lymphomas, lung cancer | In clinical use; 29 phase I–III clinical trials in progress (single and combination) | ||
| Vinflunine | Bladder, non-small-cell lung cancer, breast cancer | Phase III | ||
| Crytophycin 52 | Solid tumours | Phase III finished | ||
| Halichondrins | – | Phase I | ||
| Dolastatins | Potential vascular-targeting agent | Phase I; phase II completed | ||
| Hemiasterlins | – | Phase I | ||
| Colchicine domain | Colchicine | Non-neoplastic diseases (gout, familial mediterranean fever) | Approved in 2009 by FDA under the Unapproved Drugs Initiative[citation needed] | |
| Combretastatins | Potential vascular-targeting agent | Phase I | ||
| 2-Methoxyestradiol | – | Phase I | ||
| E7010 | Solid tumours | Phase I, II | ||
| Depolymerization inhibitors | Taxan site | Paclitaxel (Taxol) | Ovarian, breast and lung tumours, Kaposi's sarcoma; trials with numerous other tumours | In clinical use; 207 Phase I–III trials in the United States; TL00139 is in Phase I trials |
| Docetaxel (Taxotere) | Prostate, brain and lung tumours | 8 trials in the United States (Phases I–III) | ||
| Epothilone | Paclitaxel-resistant tumours | Phases I–III | ||
| Discodermolide | – | Phase I |
| Tubulin inhibitors | |||
|---|---|---|---|
| Vinca domain |  |
 |

|
| Vinblastine | Vincristine | Vinorelbine | |
 |
 |

| |
| Vinflunine | Cryptophycin 52 | Halichondrin B | |
 |
 |

| |
| Dolastatin 10 | Dolastatin 15 | Hemiasterlin A | |

| |||
| Hemiasterlin B | |||
| Colchicine domain |  |
 |

|
| Colchicine | Combretastatin | E7010 | |

| |||
| 2-Methoxyestradiol | |||
| TAXANE SITE |  |
|
-Epothilone_A.svg)
|
| Docetaxel | Paclitaxel | Epothilone A | |
-Epothilone_B.svg) |

| ||
| Epothilone B | Discodermolide | ||
Specific agents
[edit]Taxanes
[edit]Taxanes are complex terpenes produced by the plants of the genus Taxus (yews). Originally derived from the Pacific yew tree, they are now synthesized artificially. Their principal mechanism is the disruption of the cell's microtubule function by stabilizing microtubule formation. Microtubules are essential to mitotic reproduction, so through the inactivation of the microtubule function of a cell, taxanes inhibit the cell's division.
- Paclitaxel—used to treat lung cancer, ovarian cancer, breast cancer, and advanced forms of Kaposi's sarcoma.[21]
- Docetaxel—used to treat breast, ovarian, and non-small cell lung cancer.[22][23]
Vinca alkaloids
[edit]Vinca alkaloids are amines produced by the hallucinogenic plant Catharanthus roseus (Madagascar Periwinkle). Vinca alkaloids inhibit microtubule polymerization.
- Vinblastine—used to treat leukaemia, Hodgkin's lymphoma, non-small cell lung cancer, breast cancer and testicular cancer. It is also a component in a large number of chemotherapy regimens. Vinblastine and vincristine were isolated from the Madagascar periwinkle Catharanthus roseus. Madagascar traditionally used the vinca rosea to treat diabetes. In fact it has been used for centuries throughout the world to treat all kinds of ailments from wasp stings in India, to eye infections in the Caribbean. In the 1950s researchers began to analyse the plant and discovered that it contained over 70 alkaloids. Some were found to have effect on lower blood sugar levels and others act as hemostatics. The most interesting thing was that vinblastine and vincristine, were found to lower the number of white cells in blood. A high number of white cells in the blood indicates leukemia, so a new anti-cancer drug had been discovered. These two alkaloids bind to tubulin to prevent the cell from making the spindles that it needs to be able to divide. This is different from the action of taxol which interferes with cell division by keeping the spindles from being broken down. Vinblastine is mainly useful for treating Hodgkin's lymphoma, advanced testicular cancer and advanced breast cancer. Vincristine is mainly used to treat acute leukemia and other lymphomas.
- Vincristine—used to treat lymphoma, breast cancer, lung cancer, and acute lymphoblastic leukemia.[24]
- Vindesine—used to treat leukaemia, lymphoma, melanoma, breast cancer, and lung cancer.[24]
- Vinorelbine—used to treat breast cancer and non-small-cell lung cancer.[24] It was developed under the direction of the French pharmacist Pierre Poiter, who, in 1989, obtained an initial license for the dug under the brand name Navelbine. Vinorelbine is also known as vinorelbine tartrate, the drug is a semi-synthetic analogue of another cancer-fighting drug, vinblastine. Vinorelbine is included in the class of pharmaceuticals known as vinca alkaloids, and many of its characteristics mimic the chemistry and biological mechanisms of the cytotoxic drugs vincristine and vinblastine. Vinorelbine showed promising activity against breast cancer and is in clinical trial for the treatment of other types of tumors.
- Vinflunine is a novel fluorinated vinca alkaloid currently in Phase II clinical trials, which in preclinical studies exhibited superior antitumor activity to vinorelbine and vinblastine. Vinflunine block mitosis at the metaphase/anaphase transition, leading to apoptosis.[25] Vinflunine is a chemotherapy drug used to treat advanced transitional cell bladder and urothelial tract cancer. It is also called Javlor. It is licensed for people who have already had cisplatin or carboplatin chemotherapy.
Colchicine
[edit]Colchicine is an alkaloid derived from the autumn crocus (Colchicum autumnale). It inhibits mitosis by inhibiting microtubule polymerization. While colchicine is not used to treat cancer in humans, it is commonly used to treat acute attacks of gout.[26]
Colchicine is an anti-inflammatory drug that has been in continuous use for more than 3000 years. Colchicine is an oral drug, known to be used for treating acute gout and preventing acute attacks of familial Mediterranean fever (FMF). However, the use of colchicine is limited by its high toxicity in other therapies. Colchicine is known to inhibit cell division and proliferation. Early study demonstrated that colchicine disrupts the mitotic spindle. Dissolution of microtubules subsequently was shown to be responsible for the effect of colchicine on the mitotic spindle and cellular proliferation.[27]
Podophyllotoxin
[edit]Podophyllotoxin derived from the may apple plant, is used to treat viral skin infections and synthetic analogues of the molecule are used to treat certain types of cancer.
Griseofulvin
[edit]Griseofulvin, derived from a species of Penicillium is an mitotic inhibitor that is used as an antifungal drug. It inhibits the assembly of fungal microtubules
Others
[edit]- Glaziovianin A is typically isolated from the leaves of the Brazilian tree Ateleia glazioviana Baill.[28]
- Cryptophycin 52 was isolated from the blue–green algae Nostoc sp. GSV 224. The cryptophycins are a family of related depsipeptides showing highly potent cytotoxic activity. Cryptophycin 52 was originally developed as a fungicide, but was too toxic for clinical use. Later the research was focused on treating cryptophycin as a microtubule poison, preventing the formation of the mitotic spindle.[12] Cryptophycin 52 showed high potent antimitotic activity to resist spindle microtubule dynamics.[6] As well, the interest in this drug has been further arose by the discovery that cryptophycin shows reduced susceptibility to the multidrug resistance pump, and shows no reduction of activity in a number of drug-resistant cell lines.
- Halichondrin B was first isolated from Halichondria okadai, and later from the unrelated sponges Axinella carteri and Phankella carteri. Halichondrin B is a complex polyether macrolide which is synthesized and arrests cell growth at subnanomolar concentrations.[6] Halichondrin B is noncompetitive inhibitor of the binding of both vincristine and vinblastine to tubulin, suggesting the drugs bind to the vinca binding site, or a site nearby. The isolation of halichondrin B is from two unrelated genera of sponge, has led to speculate that halichondrin B is a microbial in reality, rather than sponge metabolite because sponges support a wide range of microbes. If this is the case, fermentation technologies could provide a useful supply of halichondrin B.
- Dolastatins were isolated from the sea hare Dolabella auricularia, a small sea mollusc, and thought to be the source of poison used to murder the son of Emperor Claudius of Rome in 55 A.D. Dolastatins 10 and 15 are novel pentapeptides and exhibit powerful antimitotic properties. They are cytotoxic in a number of cell lines at subnanomolar concentrations. The peptides of dolastatins 10 and 15 noncompetitively inhibit the binding of vincristine to tubulin. Dolastatin 10 is 9 times more potent than dolastatin 15 and both are more potent than vinblastin.[6] The dolastatins also enhance and stabilize the binding of colchicine to tubulin.
- Hemiasterlins were isolated from the marine sponge, Cymbastela sp. The hemiasterlins are a family of potent cytotoxic peptides. Hemiasterlin A and hemiasterlin B show potent activity against the P388 cell line and inhibit cell division by binding to the vinca alkaloid site on tubulin. Hemiasterlin A and B exhibit stronger antiproliferative activities than both the vinca alkaloids and paclitaxel.
- Combretastatins is isolated from the South African Willow, Combretum afrum. Combretastatin is one of the simpler compounds to show antimitotic effects by interaction with the colchicine binding site of tubulin, and is also one of the most potent inhibitors of colchicine binding.[6] Combretastatin is not recognized by the multiple drug resistance (MDR) pump, a cellular pump which rapidly ejects foreign molecules from the cell.[10] Combretastatin is also reported to be able to inhibit angiogenesis, a process essential for tumor growth. Except those factors, one of the disadvantage of combretastatin is the low water solubility.
- E7010 is the most active of sulfonamide antimitotic agent, which has been shown to inhibit microtubule formation by binding at the site of colchicines.[6][10] It is quite soluble in water as an acid salt. Methoxybenzene-sulfonamide showed good results against a wide range of tumor cells including vinca alkaloid resistant solid tumors. Results from animals studies indicated activity against colorectal, breast and lung cancer tissues.
- 2-Methoxyestradiol is a natural metabolite of the mammalian hormone oestradiol and is formed by oxidation in the liver. 2-methoxyestradiol is cytotoxic to several tumor cell lines, binds to the colchicine site of tubulin, inducing the formation of abnormal microtubules. 2-Methoxyestradiol exhibits potent apoptotic activity against rapidly growing tumor cells. It also has antiangiogenic activity through a direct apoptotic effect on endothelial cells.[29]
- Docetaxel, is a semi-synthetic analogue of paclitaxel, with a trade name Taxotere. Docetaxel has the minimal structure modifications at C13 side chain and C10 substitution showed more water solubility and more potency than paclitaxel. Clinical trials have shown that patients who develop hypersensitivity to paclitaxel may receive docetaxel without an allergic response.[6]
- Paclitaxel was isolated from the bark of the Pacific yew tree Taxus brevifolia Nutt. (Taxaceae). Later it was also isolated from hazelnut trees (leaves, twigs, and nuts) and the fungi living on these trees but the concentration is only around 10% of the concentration in yew trees. Paclitaxel is also known as Taxol and Onxol to be an anti-cancer drug. The drug is the first line treatment for ovarian, breast, lung, and colon cancer and the second line treatment for AIDS-related Kaposi's sarcoma. (Kaposi sarcoma is a cancer of the skin and mucous membranes that is commonly found in patients with acquired immunodeficiency syndrome, AIDS). It is so effective that some oncologists refer to the period before 1994 as the "pre-taxol" era for treating breast cancer.[30]
- Epothilones are derived from a fermenting soil bacteria, Sorangium cellulosum and it was found to be too toxic for use as an antifungal. Epothilones are microtubule stabilizing agents with a mechanism of action similar to taxanes, including suppression of microtubule dynamics, stabilization of microtubules, promotion of tubulin polymerization, and increased polymer mass at high concentrations. They induce mitotic arrest in the G2-M phase of the cell cycle, resulting in apoptosis.[5] Epothilone A and epothilone B exhibit both antifungal and cytotoxic properties. These epothilones are competitive inhibitors of the binding of paclitaxel to tubulin, exhibiting activity at similar concentrations. This finding leads to assume that the epothilones and paclitaxel adopt similar conformations in vivo. However, the epothilones are around 30 times more water-soluble than paclitaxel and more available, being easily obtained by fermentation of the parent myxobacterium and could be prepared by total synthesis. The epothilones also shows not to be recognized by multidrug resistant mechanisms, therefore it has much higher potency than paclitaxel in multidrug resistant cell lines.[10]
- Discodermolide was initially found to have immunosuppressive and antifungal activities. Discodermolide is a polyhydroxylated alketetraene lactone marine product, isolated from the Bahamian deep-sea sponge, Discodermia dissoluta, inhibited cell mitosis and induced formation of stable tubulin polymer in vitro and considered to be more effective than paclitaxel with EC50 value of 3.0 μM versus 23 μM.[6] The drug, a macrolide (polyhydroxylated lactone), is a member of a structural diverse class of compounds called polyketides with notable chemical mechanism of action. It stabilizes the microtubules of target cells, essentially arresting them at a specific stage in the cell cycle and halting cell division. It is a promising marine-derived candidate for treating certain cancers.
Limitations
[edit]Side effects
[edit]- chemotherapy-induced peripheral neuropathy, a progressive, enduring, often irreversible tingling numbness, intense pain, and hypersensitivity to cold, beginning in the hands and feet and sometimes involving the arms and legs.[31]
- stomatitis (ulceration of the lips, tongue, oral cavity)
- nausea, vomiting, diarrhea, constipation, paralytic ileus, urinary retention
- bone marrow suppression
- hypersensitivity reactions – flushing, localized skin reactions, rash (with or without) pruritus, chest tightness, back pain, dyspnea, drug fever, or chills
- musculoskeletal effects – arthralgia and/or myalgia
- severe weakness
- hypotension
- alopecia
- neurotoxicity[32]
Human factors
[edit]Limitations in anticancer therapy occur mainly due to two reasons; because of the patient's organism, or because of the specific genetic alterations in the tumor cells. From the patient, therapy is limited by poor absorption of a drug which can lead to low concentration of the active agent in the blood and small amount delivery to the tumor. Low serum level of a drug can be also caused by rapid metabolism and excretion associated with affinity to intestinal or/and liver cytochrome P450. Another reason is the instability and degradation of the drugs in gastro-intestinal environment. Serious problem is also variability between patients what causes different bioavailability after administration equal dose of a drug and different tolerance to effect of chemotherapy agents. The second problem is particularly important in treatment elderly people. Their body is weaker and need to apply lower doses, often below therapeutic level. Another problem with anticancer agents is their limited aqueous solubility what substantially reduces absorption of a drug. Problems with delivery of drags to the tumor occur also when active agent has high molecular weight which limits tissue penetration or the tumor has large volume prevent for penetration.[4][33]
Drug resistance
[edit]Multidrug resistance is the most important limitation in anticancer therapy. It can develop in many chemically distinct compounds. Until now, several mechanisms are known to develop the resistance. The most common is production of so-called "efflux pumps". The pumps remove drugs from tumor cells which lead to low drug concentration in the target, below therapeutic level. Efflux is caused by P-glycoprotein called also the multidrug transporter. This protein is a product of multidrug resistance gene MDR1 and a member of family of ATP-dependent transporters (ATP-binding cassette). P-glycoprotein occurs in every organism and serves to protect the body from xenobiotics and is involved in moving nutrients and other biologically important compounds inside one cell or between cells. P-glycoprotein detects substrates when they enter the plasma membrane and bind them which causes activation of one of the ATP-binding domains. The next step is hydrolysis of ATP, which leads to a change in the shape of P-gp and opens a channel through which the drug is pumped out of the cell. Hydrolysis of a second molecule of ATP results in closing of the channel and the cycle is repeated. P-glycoprotein has affinity to hydrophobic drugs with a positive charge or electrically neutral and is often over-expressed in many human cancers. Some tumors, e.g. lung cancer, do not over-express this transporter but also are able to develop the resistance. It was discovered that another transporter MRP1 also work as the efflux pump, but in this case substrates are negatively charged natural compounds or drugs modified by glutathione, conjugation, glycosylation, sulfation and glucuronylation. Drugs can enter into a cell in few kinds of ways. Major routes are: diffusion across the plasma membrane, through receptor or transporter or by the endocytosis process. Cancer can develop the resistance by mutations to their cells which result in alterations in the surface of cells or in impaired endocytosis. Mutation can eliminate or change transporters or receptors which allows drugs to enter into the tumor cell. Other cause of drug resistance is a mutation in β tubulin which cause alterations in binding sites and a given drug cannot be bound to its target. Tumors also change expression isoforms of tubulin for these ones, which are not targets for antimitotic drugs e.g. overexpress βIII-tubulin. In addition tumor cells express other kinds of proteins and change microtubule dynamic to counteract effect of anticancer drugs. Drug resistance can also develop due to the interruption in therapy.[4][7][8][33]
Others
[edit]- Marginal clinical efficacy – often compounds show activity in vitro but do not have antitumor activity in clinic.[34]
- Poor water solubility of drugs which need to be dissolved in polyoxyethylated castor oil or polysorbate what cause hypersensitivity reactions. It has been suggested this solvents can also reduce delivery of the drugs to target cells.[12][35]
- Bioavailability[36]
- Dose limit – higher doses cause high toxicity and long-term use lead to cumulative neurotoxicity and hematopoietic toxicity.[12]
- Neuropathy which is significant side effect can develop at any time in therapy and require an interruption of treatment. After symptoms have resolved therapy can be started again but the break allow tumor for develop of resistance.[20]
- Poor penetration through the blood–brain barrier.[20]
Discovery and development
[edit]The first known compound which binds to tubulin was colchicine, it was isolated from the autumn crocus, Colchicum autumnale, but it has not been used for cancer treatment. First anticancer drugs approved for clinical use were Vinca alkaloids, vinblastine and vincristine in the 1960s.
They were isolated from extracts leaves of the Catharanthus roseus (Vinca rosea) plant at the University of Western Ontario in 1958.[5] First drug belong to the taxanes and paclitaxel, discovered in extracts from the bark of the yew tree, Taxus brevifolia, in 1967 by Monroe Wall and Mansukh Wani but, its tubulin inhibition activity was not known until 1979.
Yews trees are poor source of active agents that limited the development of taxanes for over 20 years until discover the way of synthesis.[5] In December 1992 paclitaxel was approved to use in chemotherapy.[37]
Future drug development
[edit]Because of numerous adverse effect and limitations in use, new drugs with better properties are needed. Especially are desired improvements in antitumor activity, toxicity profile, drug formulation and pharmacology.[35] Currently have been suggested few approaches in development of novel therapeutic agents with better properties
- Discovery agents which are not a substrate for efflux pump or modifications of drugs in toward lower affinity to transporting proteins. Discover P-glycoprotein inhibitors with higher affinity to the transporter then drugs, is next approach. For improving oral bioavailability is suggested co-administration of P-gp and cytochrome inhibitors with anticancer drugs.[20][35]
- Development of inhibitors that have their binding site in α-tubulin. This part of tubulin dimer remains unused because all currently use drugs bind to the β-tubulin. Research in this field can open new opportunity in treatment and provide new class of inhibitors.
- One of the targets for anticancer drugs can be tumor vasculature. The advantage in this case is relatively easy access of therapeutic agents to the target. It is known that some compounds can inhibit the formation of new blood vessels (inhibit the process of angiogenesis) or shut down existing ones. Tumor cells die very fast after cutting off the oxygen supply what suggest these agents are especially interesting. What more, it seems the agents act only with tumor vasculature and do not interact with normal tissues. The mechanisms is not known but has been suggested that the reason are differences between young tissue of tumor and mature tissue of normal vasculature. Antivascular agents are similar to colchicine and bind to the colchicine binding site on β-tubulin so development of novel agents acting with colchicine binding site (which is not used by any of currently approved drugs) seems to be a promising approach.[5]
- Therapy with combination of two or more drugs which have various binding sites and/or different mechanism of action but have non overlapping adverse effects. This would allow use of drugs in low concentration what reduce strength of side effects associated with high doses of anticancer agents. Better efficiency might be also a result of maintenance low concentrations of drugs for long period instead of drastic changes in the amount of administered drugs.[8][12]
- Liposomes and polymer-bound drugs comprise promising improvements in delivery system. Liposomes allow for delivery considerable amounts of drag to the tumor without toxic effect in normal tissues and slowly release drugs what result in prolongation of pharmaceutical action. Similar properties have drugs bound to polymer. In addition, use of water-soluble polymers allow hydrophilic anticancer agents become soluble. The nature of polymer-drug linkage can be designed to be stable in normal tissues and break down in tumor environment, which is more acidic. This approach allow for release active agent exactly in the target.[36]
- Discover new compounds active against drug-resistant cancers with different mechanism than drugs have been already known.
- Elucidation of all resistance mechanisms and design drugs which avoid it.[12]
This section needs to be updated. (December 2016) |
See also
[edit]- Medicinal molds
- Tubulin
- Microtubule
- Cancer
- Chemotherapy
- Drug design
- Vinblastine
- Vincristine
- Vinorelbine
- Vinflunine
- Cryptophycin
- Halichondrin B
- Colchicine
- Combretastatins
- 2-Methoxyestradiol
- Docetaxel
- Paclitaxel
- Epothilones
- Discodermolide
- eribulin, a newer agent
References
[edit]- ^ a b "What Are the Different Types of Chemotherapy Drugs?". American Cancer Society. Archived from the original on 17 July 2007. Retrieved 5 August 2007.
- ^ "Definition of mitotic inhibitor". National Cancer Institute. Archived from the original on 13 August 2024. Retrieved 5 August 2007.
- ^ "Treatment Options: Mitotic Inhibitors". Drug Digest. Archived from the original on 16 February 2007. Retrieved 5 August 2007.
- ^ a b c d e f Perez, E. A. (2009). "Microtubule inhibitors: Differentiating tubulin-inhibiting agents based on mechanisms of action, clinical activity, and resistance". Molecular Cancer Therapeutics. 8 (8): 2086–95. doi:10.1158/1535-7163.MCT-09-0366. PMID 19671735.
- ^ a b c d e f g h Jordan, M. (2012). "Mechanism of Action of Antitumor Drugs that Interact with Microtubules and Tubulin". Current Medicinal Chemistry. Anti-Cancer Agents. 2 (1): 1–17. doi:10.2174/1568011023354290. PMID 12678749.
- ^ a b c d e f g h i j Islam, Mohd.; Iskander, Magdy (2004). "Microtubulin Binding Sites as Target for Developing Anticancer Agents". Mini-Reviews in Medicinal Chemistry. 4 (10): 1077–104. doi:10.2174/1389557043402946. PMID 15579115.
- ^ a b c d Pellegrini, Federico; Budman, Daniel R (2005). "Review: Tubulin Function, Action of Antitubulin Drugs, and New Drug Development". Cancer Investigation. 23 (3): 264–73. doi:10.1081/CNV-200055970. PMID 15948296. S2CID 45866448.
- ^ a b c d e f Jordan, Mary Ann; Wilson, Leslie (2004). "Microtubules as a target for anticancer drugs". Nature Reviews Cancer. 4 (4): 253–65. doi:10.1038/nrc1317. PMID 15057285. S2CID 10228718.
- ^ TitoFojo, The role of microtubules in Cell Biology, Neurobiology and Oncology, Humana Press.[page needed]
- ^ a b c d Jordan, Allan; Hadfield, John A.; Lawrence, Nicholas J.; McGown, Alan T. (1998). "Tubulin as a target for anticancer drugs: Agents which interact with the mitotic spindle". Medicinal Research Reviews. 18 (4): 259–96. doi:10.1002/(SICI)1098-1128(199807)18:4<259::AID-MED3>3.0.CO;2-U. PMID 9664292. S2CID 32194348.
- ^ Bhalla, Kapil N (2003). "Microtubule-targeted anticancer agents and apoptosis". Oncogene. 22 (56): 9075–86. doi:10.1038/sj.onc.1207233. PMID 14663486.
- ^ a b c d e f Morris, P. G.; Fornier, M. N. (2008). "Microtubule Active Agents: Beyond the Taxane Frontier". Clinical Cancer Research. 14 (22): 7167–72. doi:10.1158/1078-0432.CCR-08-0169. PMID 19010832.
- ^ a b Chen, Jing; Liu, Tao; Dong, Xiaowu; Hu, Yongzhou (2009). "Recent Development and SAR Analysis of Colchicine Binding Site Inhibitors". Mini-Reviews in Medicinal Chemistry. 9 (10): 1174–90. doi:10.2174/138955709789055234. PMID 19817710.
- ^ Abal, M.; Andreu, J.; Barasoain, I. (2003). "Taxanes: Microtubule and Centrosome Targets, and Cell Cycle Dependent Mechanisms of Action". Current Cancer Drug Targets. 3 (3): 193–203. doi:10.2174/1568009033481967. PMID 12769688.
- ^ Fang, W.-; Liang, X.- (2005). "Recent Progress in Structure Activity Relationship and Mechanistic Studies of Taxol Analogues". Mini-Reviews in Medicinal Chemistry. 5 (1): 1–12. doi:10.2174/1389557053402837. PMID 15638787.
- ^ Lixin Zhang, Arnold L. Demain (2005), Natural products: drug discovery and therapeutic medicine.Natural products: drug discovery and therapeutic medicine Archived 30 October 2023 at the Wayback Machine[page needed]
- ^ Hamel, Ernest (1996). "Antimitotic natural products and their interactions with tubulin". Medicinal Research Reviews. 16 (2): 207–31. doi:10.1002/(SICI)1098-1128(199603)16:2<207::AID-MED4>3.0.CO;2-4. PMID 8656780. S2CID 647015. Archived from the original on 22 August 2020. Retrieved 21 January 2024.
- ^ Kingston, David G. I. (2009). "Tubulin-Interactive Natural Products as Anticancer Agents(1)". Journal of Natural Products. 72 (3): 507–15. doi:10.1021/np800568j. PMC 2765517. PMID 19125622.
- ^ Cragg, Gordon M.; Newman, David J. (2004). "A Tale of Two Tumor Targets: Topoisomerase I and Tubulin. The Wall and Wani Contribution to Cancer Chemotherapy†". Journal of Natural Products. 67 (2): 232–44. doi:10.1021/np030420c. PMID 14987065.
- ^ a b c d Kuppens, Isa (2006). "Current State of the Art of New Tubulin Inhibitors in the Clinic". Current Clinical Pharmacology. 1 (1): 57–70. doi:10.2174/157488406775268200. PMID 18666378.
- ^ Saville, M. W.; Lietzau, J.; Pluda, J. M.; Wilson, W. H.; Humphrey, R. W.; Feigel, E.; Steinberg, S. M.; Broder, S.; Yarchoan, R.; Odom, J.; Feuerstein, I. (1995). "Treatment of HIV-associated Kaposi's sarcoma with paclitaxel". Lancet. 346 (8966): 26–28. doi:10.1016/S0140-6736(95)92654-2. PMID 7603142. Archived from the original on 26 June 2019. Retrieved 5 July 2019.
- ^ Lyseng-Williamson, K. A.; Fenton, C. (2005). "Docetaxel: A Review of its Use in Metastatic Breast Cancer". Drugs. 65 (17): 2513–2531. doi:10.2165/00003495-200565170-00007. PMID 16296875.
- ^ Clarke, S. J.; Rivory, L. P. (1999). "Clinical Pharmacokinetics of Docetaxel". Clinical Pharmacokinetics. 36 (2): 99–114. doi:10.2165/00003088-199936020-00002. PMID 10092957.
- ^ a b c "Vincristine (Oncovin)". Archived from the original on 29 June 2007. Retrieved 5 August 2007.
- ^ Okouneva, Tatiana; Hill, Bridget T.; Wilson, Leslie; Jordan, Mary Ann (2003). "The Effects of Vinflunine, Vinorelbine, and Vinblastine on Centromere Dynamics". Molecular Cancer Therapeutics. 2 (5): 427–36. PMID 12748304. Archived from the original on 13 August 2024. Retrieved 21 January 2024.
- ^ Lu Y, Chen J, Xiao M, Li W, Miller DD (November 2012). "An overview of tubulin inhibitors that interact with the colchicine binding site". Pharm Res. 29 (11): 2943–71. doi:10.1007/s11095-012-0828-z. PMC 3667160. PMID 22814904.
- ^ Molad, Yair (2002). "Update on colchicine and its mechanism of action". Current Rheumatology Reports. 4 (3): 252–6. doi:10.1007/s11926-002-0073-2. PMID 12010611. S2CID 4507579.
- ^ Writer, GEN Staff (29 June 2016). "Fighting Cancer with a Pinch of Parsley and Dill". GEN – Genetic Engineering and Biotechnology News. Retrieved 26 October 2023.
- ^ Lakhani, Nehal J.; Sarkar, Mohamadi A.; Venitz, Jurgen; Figg, William D. (2003). "2-Methoxyestradiol, a Promising Anticancer Agent". Pharmacotherapy. 23 (2): 165–72. doi:10.1592/phco.23.2.165.32088. PMID 12587805. S2CID 1541302. Archived from the original on 25 June 2023. Retrieved 21 January 2024.
- ^ http://www.paclitaxel.org/[full citation needed]
- ^ del Pino BM (23 February 2010). "Chemotherapy-induced Peripheral Neuropathy". NCI Cancer Bulletin. p. 6. Archived from the original on 11 December 2011.
- ^ Hazardous Substances Data Bank (HSDB) http://toxnet.nlm.nih.gov Archived 11 June 2019 at the Wayback Machine[full citation needed]
- ^ a b Gottesman, Michael M. (2002). "Mechanisms of cancer drug resistance". Annual Review of Medicine. 53: 615–27. doi:10.1146/annurev.med.53.082901.103929. PMID 11818492. Archived from the original on 13 August 2024. Retrieved 21 January 2024.
- ^ Ivachtchenko, Alexandre; Kiselyov, Alex; Tkachenko, Sergey; Ivanenkov, Yan; Balakin, Konstantin (2007). "Novel Mitotic Targets and Their Small-Molecule Inhibitors". Current Cancer Drug Targets. 7 (8): 766–84. doi:10.2174/156800907783220499. PMID 18220536.
- ^ a b c Attard, Gerhardt; Greystoke, Alastair; Kaye, Stan; De Bono, Johann (2006). "Update on tubulin-binding agents". Pathologie Biologie. 54 (2): 72–84. doi:10.1016/j.patbio.2005.03.003. PMID 16545633.
- ^ a b Terwogt, Jetske M.Meerum; Schellens, Jan H.M.; Huinink, Wim W.ten Bokkel; Beijnen, Jos H. (1999). "Clinical pharmacology of anticancer agents in relation to formulations and administration routes". Cancer Treatment Reviews. 25 (2): 83–101. doi:10.1053/ctrv.1998.0107. PMID 10395834.
- ^ Gordaliza, M. (2008). "Natural products as leads to anticancer drugs". Clinical and Translational Oncology. 9 (12): 767–76. doi:10.1007/s12094-007-0138-9. PMID 18158980. S2CID 19282719.